Advancements and Opportunities in Targeted Protein Degradation: Unveiling Novel Therapeutic Pathways
Abstract
The revolution in cancer genomics has provided a wealth of promising and challenging targets for drug discovery. In the field of Targeted Protein Degradation (TPD), small molecules are used to reprogram the protein homeostasis system, disrupting the targeted proteins essential for the process. This article provides a high-level assessment of the advancements in this field and offers a review of the steps necessary to realize the full potential of this novel therapeutic approach.
The emergence of the TPD field
Manipulating proteins using chemical methods in the intracellular environment is a cornerstone of chemical biology and drug discovery. Given that nature often regulates biochemical processes through scaffold proteins or co-sequestration complexes to induce proximity, the first well-established example of such processes, often termed “molecular glues” or chemical induced proximity (CIP), is the macrolide natural product FK506 (Figure 1A). It simultaneously binds to the protein FK506, protein 12 (FKBP12), and calcineurin. In the three decades since the pioneering work on FK506, many other inducers of proximity, such as FK1012 (Fig 1A), a homodimer of FK506, have been discovered to reconnect cellular circuits. Subsequently, to circumvent the use of peptide ligands, numerous bivalent degraders based on von Hippel-Lindau (VHL) ligands were developed, recruiting the CRL2VHL E3 ubiquitin ligase. Following this, based on the mechanism of action (MOA) study of the IMiD drug thalidomide (Fig 1B), a series of independently developed bifunctional degraders recruiting CRBN using IMiDs and IMiD derivatives emerged. Building upon this, the development of degradation tag fusion strategies allowed for faster functional loss studies. In summary, the early discoveries of target degradation can yield pharmacological effects, small molecule ligands of E3 ubiquitin ligases can be rationally employed for targeted degradation, thus laying the foundation for the subsequent surge of interest in Targeted Protein Degradation (TPD).
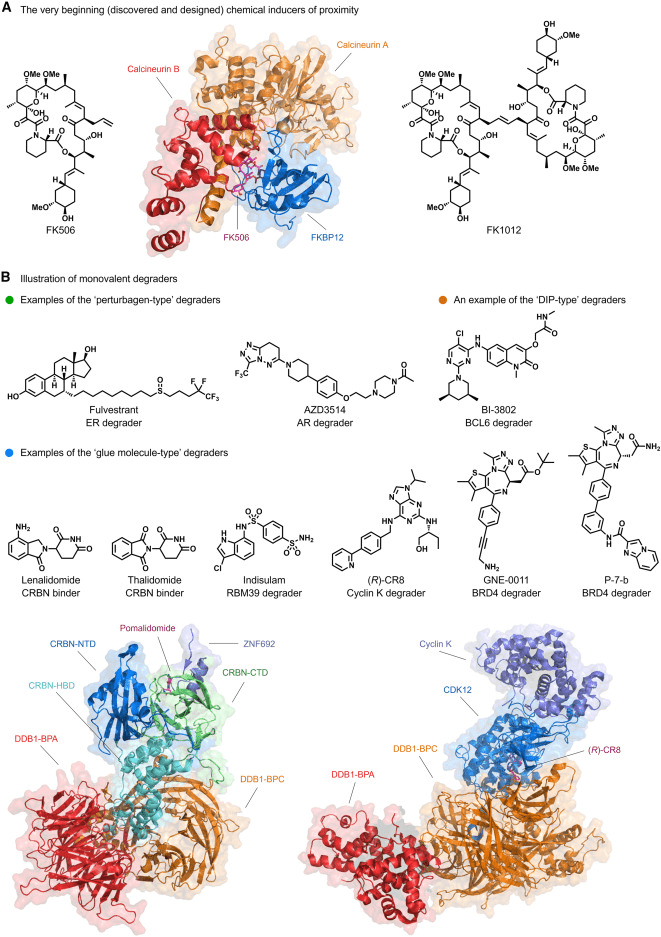
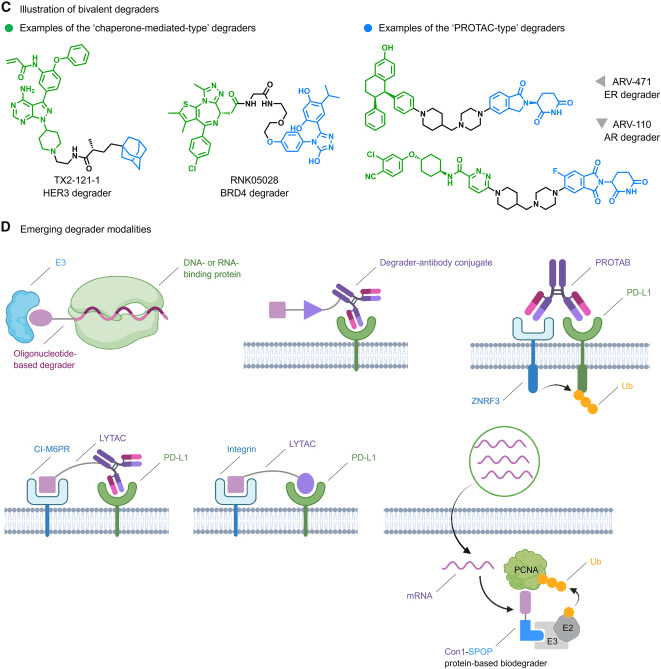
Fig 1. Schematic representation of proximity-induced protein degraders [1].
The primary forms of Targeted Protein Degradation (TPD) – monovalent and bivalent small molecule degraders.
In essence, monovalent and bivalent small molecule degraders represent the primary forms of Targeted Protein Degradation (TPD). Monovalent degraders consist of only one binding ligand. They can act as perturbations to drive the target protein into a conformational state that is susceptible to degradation. Alternatively, they can function as glue molecules that preferentially bind to either the target protein or an E3 ligase, modifying their surfaces and inducing protein-protein interactions (PPIs) with complementary partners. Notably, perturbation-based degraders like fulvestrant for hormone receptor (HR)-positive advanced cancer (Fig1B) and the molecular glue-type degrader Indisulam targeting RNA binding motif protein 39 (RBM39) (Fig 1B) are representative examples. However, the discovery of monovalent degraders has lagged behind bivalent ones due to the lack of rational discovery strategies.
Bivalent degraders are composed of two binding ligands connected by a linker, with each ligand binding to the target protein and an E3 ligase, respectively, to achieve spatial proximity. Representative examples include chimeric compounds mediated by recruiting proteins (Fig 1C), Proteolysis-Targeting Chimeras (PROTACs) (Fig 1C), and Lysosome-Targeting Chimeras (LYTACs) (Fig 1D). Among them, the estrogen receptor (ER) degrader ARV-471 (Fig 1C) has entered Phase III trials. The following sections provide a detailed description of the steps towards realizing the full potential of the TPD paradigm.
Translation opportunities in TPD
Overview of pure play and partnerships on the TPD track
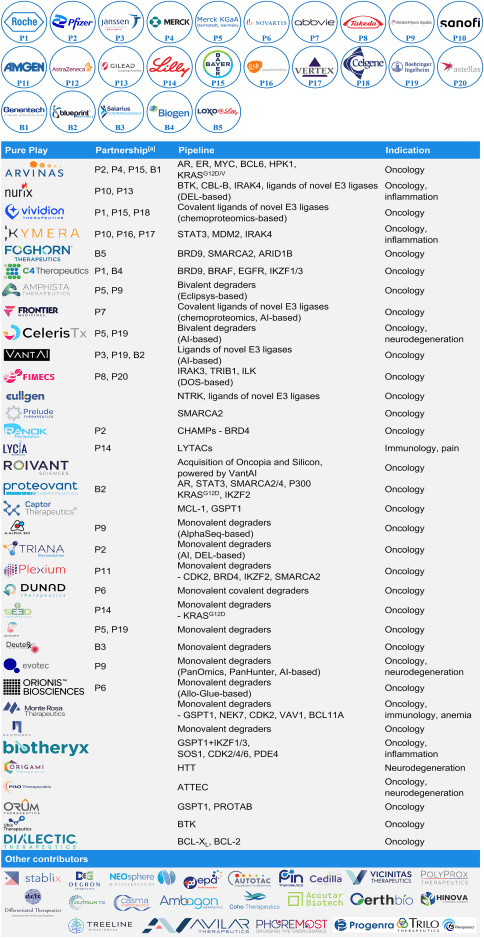
Since the establishment of companies dedicated to TPD such as Nurix and Arvinas, the field of TPD has attracted substantial investments, giving rise to numerous new companies and partnerships (Table 1). Firstly, in terms of targets, some companies choose to focus on clinically validated targets. For example, Arvinas’ ARV-110 (Fig 1C) and ARV-471, both PROTAC molecules, are among the first batch of PROTACs to enter Phase II trials, and the field eagerly awaits their outcomes. Secondly, in terms of indications, oncology remains the most popular therapeutic area, while other fields such as neurodegenerative diseases, inflammation, and immunology are just beginning to be explored. Finally, the wave of AI-supported drug discovery is making its way into the TPD field. Companies like Celeris, VantAI, Roivant, Triana, and Evotec are actively influencing and accelerating the optimization of bivalent degraders, identifying new E3 ubiquitin ligase ligands, and discovering monovalent degraders. Overall, the disease target combinations in the product pipelines of TPD companies reflect the trends in degradation drug discovery and development.
Table 1. Selected TPD contributors and their pipelines [1].
Unveiling the Opportunities Hidden Beneath the Iceberg
Given the extensive attention on the potential of TPD, its combined applications with fields such as synthetic biology, protein engineering, and nucleic acid regulation are rapidly diversifying. For instance, degrader-antibody conjugates (Fig 1D) originating from antibody-drug conjugates (ADCs) and bispecific antibody degraders targeting Programmed Cell Death Ligand 1 (PD-L1), EGFR, and Insulin-like Growth Factor 1 Receptor (IGF1R) (Fig 1D) are being developed. Furthermore, protein-based biodegraders (Fig 1D) have demonstrated the ability to effectively clear potent tumor co-targets like Proliferating Cell Nuclear Antigen (PCNA) following the delivery of degrader mRNA using lipid nanoparticles, significantly expanding the scope of degradable genomes.
In the realm of anti-infective applications, TPD approaches have shown promise for the X protein of Hepatitis B Virus (HBV) and the NS3/4A protease of Hepatitis C Virus (HCV). Apart from inducing ubiquitination, emerging proximity-induced modes hold potential to induce other post-translational modifications (PTMs) like deubiquitination, dephosphorylation, phosphorylation, and acetylation, to target protein stability, inactivation, activation, and localization. In summary, these innovative endeavors further propel the development of this field, creating additional avenues for transformation.
Implementing the “5Rs” Principle for Successful Translation
In 2014, AstraZeneca introduced a valuable five-dimensional framework, known as the “5Rs”: Right Target, Right Tissue, Right Safety, Right Patient, and Right Commercial Potential, to guide the development of small molecule drugs. For TPD, one of the most crucial lessons from the 5Rs is to identify the optimal approach, where degradation strategies outperform traditional inhibition strategies. The development of a degrader for Signal Transducer and Activator of Transcription 3 (STAT3), named SD-36 (Fig 2), used to treat blood malignancies driven by STAT3 signaling, serves as an excellent example. As monomeric STAT3 still retains transcription factor activity, STAT3 inhibitors are only partially effective. Hence, degradation methods are favored over inhibition strategies. In the context of TPD, adding another layer of target tissue specificity involves pairing cell-specific or tissue-specific E3 ubiquitin ligases. The development of BCL-XL degraders like DT2216 and 753b is a notable instance, addressing platelet toxicity associated with BCL-XL inhibition (Fig 2). In essence, tailoring degradation in the disease context will contribute to successful translation.
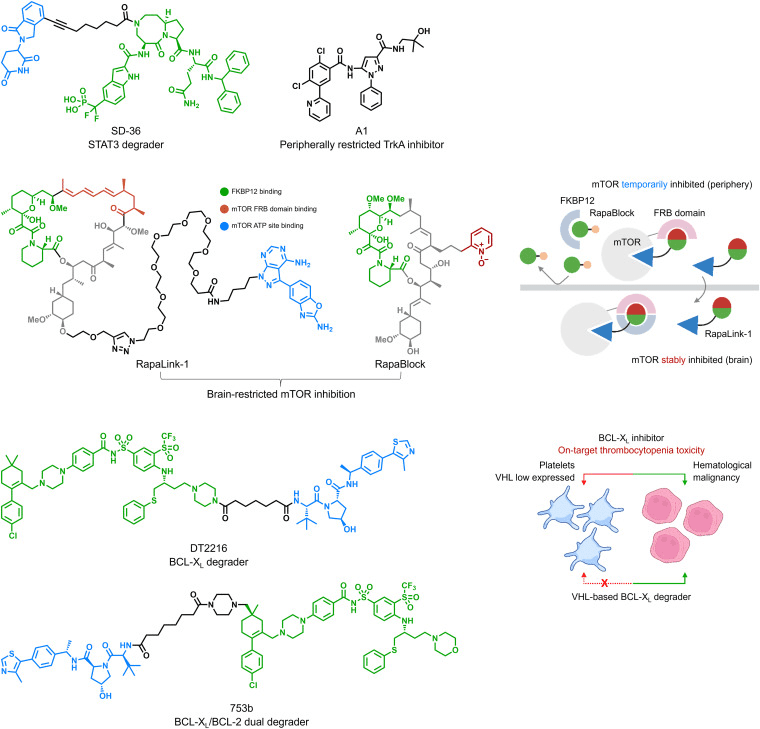
Fig 2. Chemical structures of SD-36, DT2216, and 753b [1].
Breaking the Constraints of Lipinski’s Rule of 5: Synergistic Uptake Transporters
Oral administration remains the most convenient and cost-effective drug delivery route. As mentioned earlier, the development of oral bivalent drugs is far more challenging than oral inhibitors. Generally guided by Lipinski’s Rule of Five (Ro5), in the TPD field, Ro5 does not apply to compounds that serve as substrates for biological uptake transporters (Figure 3A). The first example is the inhibitor of Hepatitis C Virus Non-Structural Protein 5A (HCV-ns5a). Since the disclosure of daclatasvir, a variety of inhibitors including ledipasvir, elbasvir, velpatasvir, ombitasvir, and pibrentasvir (Fig 3B) have been developed and approved for treating chronic HCV. According to Ro5, these high-dose inhibitors are expected to have poor oral bioavailability. However, the discovery of L-Pro-L-Val dipeptide moieties present in these molecules, recognized by transporters, ensures high plasma exposure through oral administration.
The second example pertains to the BCL-2/BCL-XL dual inhibitor navitoclax (ABT-263) and the BCL-2 inhibitor venetoclax (ABT-199) (Fig 3C), used to treat relapsed or refractory chronic lymphocytic leukemia. Both molecules are suitable for oral administration, even though navitoclax and venetoclax violate Ro5. Research has shown that intestinal lymphatic transport plays a significant role in their absorption, potentially increasing systemic exposure by avoiding first-pass liver effects. In conclusion, utilizing intestinal and brain uptake transporters for the absorption of bivalent degrader drugs might represent a unique direction to address challenges in oral bioavailability and central nervous system exposure.
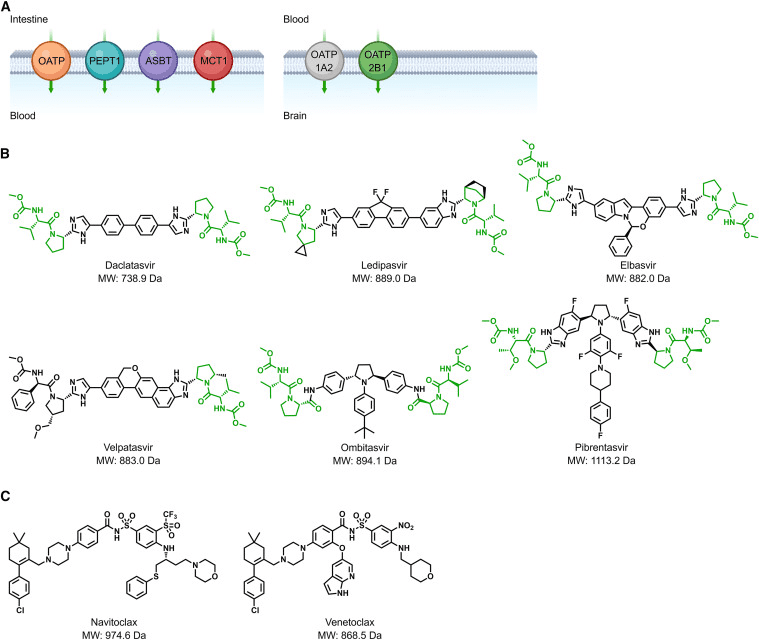
Fig 3. Schematic representation of transporter-assisted drug absorption [1].
In conclusion, with the remarkable progress achieved in the field of targeted protein degradation, a multitude of therapeutic hypotheses have already transitioned or are in the process of transitioning into clinical research, heralding the imminent era of degrader therapeutics. In this context, we endeavor to timely present our observations and thoughts, highlighting opportunities and approaches for translation, with the aim of proposing steps that could unlock the full potential of degradation therapeutics.
Reference
- Teng, M., & Gray, N. S. (2023). The rise of degrader drugs. Cell chemical biology, 30(8), 864–878.
- Stanton, B. Z., Chory, E. J., & Crabtree, G. R. (2018). Chemically induced proximity in biology and medicine. Science (New York, N.Y.), 359(6380), eaao5902.
- Sakamoto, K. M., Kim, K. B., Kumagai, A., Mercurio, F., Crews, C. M., & Deshaies, R. J. (2001). Protacs: chimeric molecules that target proteins to the Skp1-Cullin-F box complex for ubiquitination and degradation. Proceedings of the National Academy of Sciences of the United States of America, 98(15), 8554–8559.
- Buckley, D. L., Van Molle, I., Gareiss, P. C., Tae, H. S., Michel, J., Noblin, D. J., Jorgensen, W. L., Ciulli, A., & Crews, C. M. (2012). Targeting the von Hippel-Lindau E3 ubiquitin ligase using small molecules to disrupt the VHL/HIF-1α interaction. Journal of the American Chemical Society, 134(10), 4465–4468.
- Nowak, R. P., & Jones, L. H. (2021). Target Validation Using PROTACs: Applying the Four Pillars Framework. SLAS discovery : advancing life sciences R & D, 26(4), 474–483.