Advances in FGFR: Cancer targets, therapy, and signaling elucidation
Abstract
Fibroblast growth factor receptors (FGFRs) are cell surface receptors that play critical roles in cell growth, differentiation, and tissue repair. They are activated by binding to fibroblast growth factors (FGFs), triggering signaling pathways involved in development and disease. Dysregulation of FGFR signaling is implicated in various cancers and developmental disorders. Targeting FGFRs with inhibitors is a promising approach in cancer therapy, with several inhibitors in clinical trials. Understanding FGFR signaling is essential for developing treatments that can modulate these pathways effectively.
What is FGFR?
The fibroblast growth factor receptor (FGFR) is a transmembrane receptor tyrosine kinase that plays a crucial role in various biological processes. FGFR comprises four subtypes, FGFR1-4, and is activated by binding to fibroblast growth factors (FGFs). This binding induces receptor dimerization and phosphorylation, initiating downstream signaling pathways involved in cell proliferation, migration, survival, and angiogenesis.
Genetic mutations in FGFR, such as amplification, mutation, or rearrangement, can lead to the overexpression or activation of FGFR, promoting tumor growth and metastasis. Consequently, FGFR inhibitors have been developed and are currently used in clinical practice to treat patients with advanced FGFR gene mutations in cholangiocarcinoma or urothelial cancer. Examples of these inhibitors include erdafitinib, pemigatinib, infigratinib, and futibatinib.
Furthermore, FGFR inhibitors have shown efficacy in treating other solid tumors, such as gastrointestinal, lung, and gynecological tumors. These inhibitors represent a significant advancement in targeted cancer therapy, offering new treatment options for patients with FGFR-related malignancies.
Current status of FGFR targeted therapy
Fibroblast growth factor (FGF) plays a crucial role in fetal development and in maintaining tissue and systemic homeostasis by signaling through FGF receptors (FGFR1-4). However, mutations, rearrangements, or fusions involving FGFR genes can lead to tumorigenesis. FGFR fusions can be categorized into type I (non-receptor type, caused by N-terminal fusion partner substitution, e.g., BCR-FGFR1 and ZYM2-FGFR1), type IIa (receptor type, N-terminal fusion partner substitution, e.g., FN1-FGFR1, KLK2-FGFR2), or type IIb (receptor type, C-terminal fusion partner substitution, e.g., FGFR2-BICC1, FGFR3-TACC3).
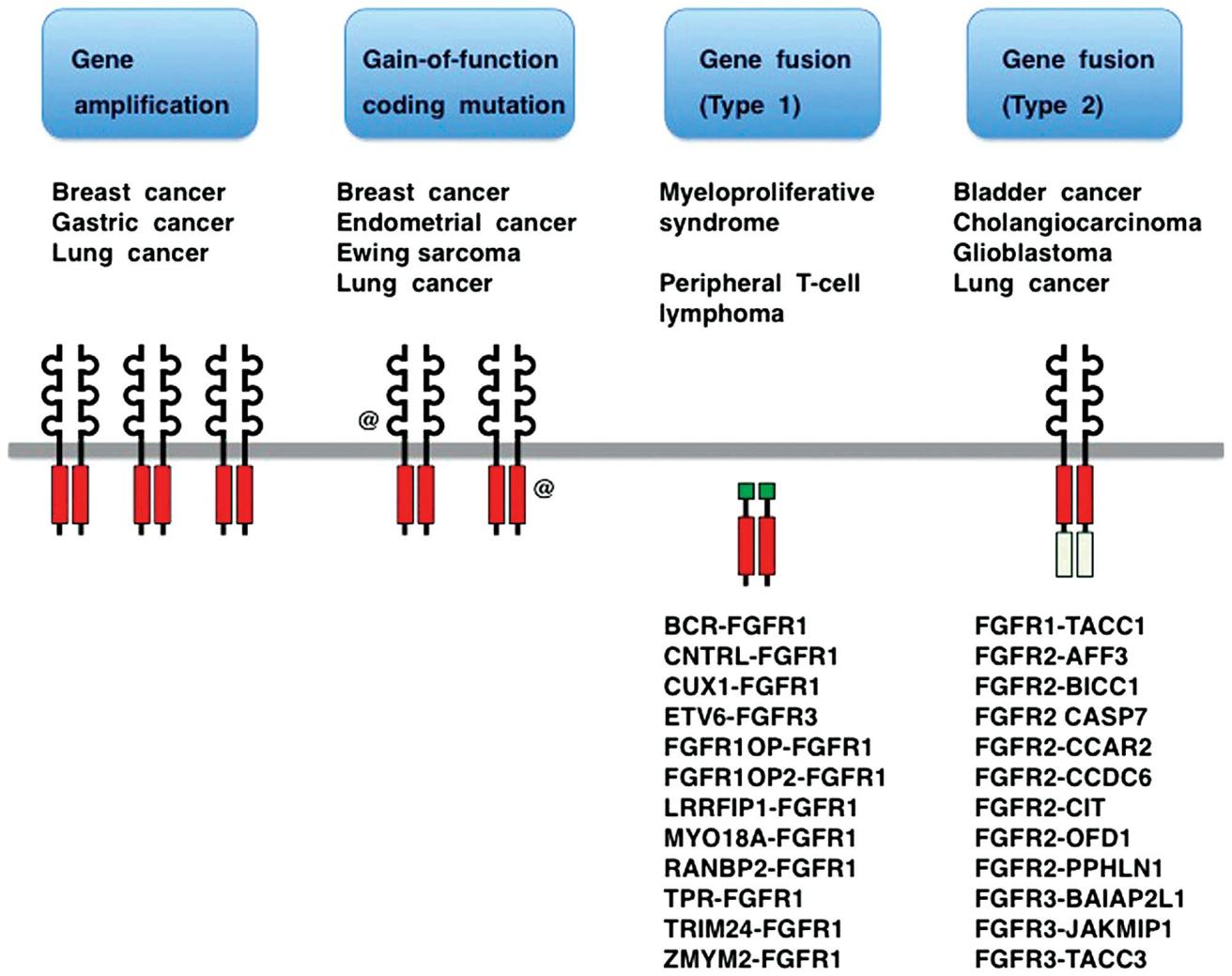
Figure 1 – Fibroblast growth factor receptor (FGFR) alterations in human cancer.
In solid tumors, FGFR alterations are observed, such as FGFR1 amplification in non-small cell lung cancer (20%), FGFR1/2 amplification in breast cancer (7-23%), FGFR3 mutations in urothelial carcinoma (10-60%), or FGFR3 fusions (6%), FGFR2 fusions in intrahepatic cholangiocarcinoma (10-20%), FGFR2 mutations in endometrial cancer (12%), and FGFR2 amplification in gastric cancer (5-10%). Approximately 7.0% of unselected cancer patients exhibit FGFR gene alterations.
Several FGFR-targeting drugs have been developed, including pan-FGFR inhibitors (erdafitinib and futibatinib), FGFR1/2/3 inhibitors (infigratinib and pemigatinib), and more specific drugs, some of which are in clinical use. Erdafitinib is approved for urothelial cancer patients with FGFR2/3 mutations, while futibatinib and pemigatinib are approved for cholangiocarcinoma patients with FGFR2 fusions or rearrangements. However, the clinical benefit of these drugs is limited by hyperphosphatemia from off-target FGFR1 inhibition and the emergence of drug-resistant mutations in FGFR. Next-generation inhibitors like lirafugratinib and LOXO-435, and the FGFR2-specific antibody bemarituzumab, promise to reduce hyperphosphatemia risk and overcome certain resistance mutations.
FGFR structure and signaling pathways
Fibroblast growth factors (FGFs) are characterized by conserved core domains with globular or atypical β-trefoil structures. They can be classified into typical FGFs (FGF1-10, FGF16-18, FGF20, and FGF22), endocrine FGFs (FGF19/FGF15, FGF21, and FGF23), and FGF homologous factors (FGF11-14). Full-length FGF receptors (FGFR1-4) are type I transmembrane proteins with three extracellular immunoglobulin-like domains and an intracellular tyrosine kinase domain.
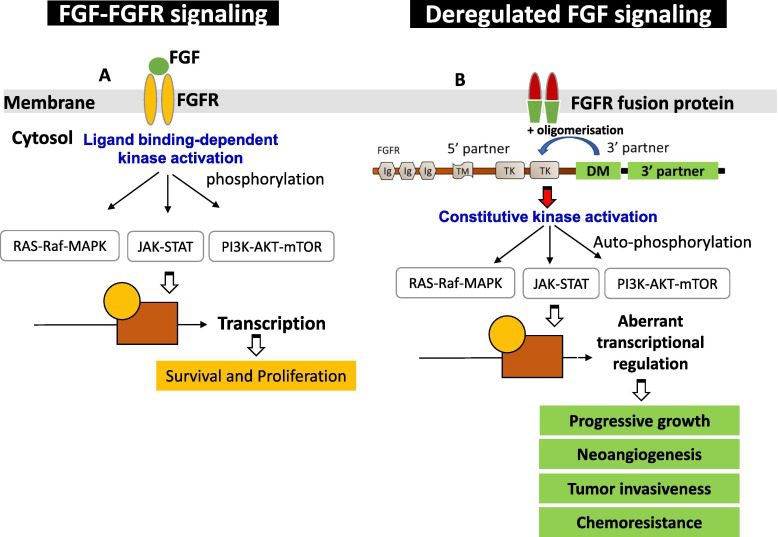
Figure 2 -FGF-FGFR signaling pathway
Canonical FGF signaling occurs through FGFRs in conjunction with the heparan sulfate cofactor, while endocrine FGF signaling requires the klotho co-receptors (KLA and KLB) in addition to the heparan sulfate cofactor. FGF-FGFR signaling plays critical roles in cell survival, proliferation, metabolism, migration, and differentiation, including coordinating fetal development and maintaining tissue and systemic homeostasis. However, aberrant FGF-FGFR signaling can drive tumorigenesis.
FGFR activation can occur through ligand-dependent dimerization or pathological changes, leading to endogenous kinase activation by releasing autoinhibitory mechanisms. Upon activation, FGFR stimulates downstream signaling pathways, including FRS2-dependent activation of PI3K-AKT, RAS-ERK, and phospholipase Cγ (PLCγ)-dependent diacylglycerol (DAG)-PKC and IP3-Ca2+ signaling.
In cancer, FGFR alterations promote tumorigenesis through abnormal FGFR signaling. FGFR amplification results in FGFR overexpression and overactivation of downstream pathways. FGFR fusion or rearrangement leads to fusion partner-mediated FGFR dimerization and abnormal downstream signaling. FGFR mutations in the extracellular region or transmembrane domain alter specificity or affinity for FGF ligands, leading to ligand-independent dimerization. Mutations in the tyrosine kinase domain constitutively activate FGFR by disrupting the autoinhibitory mechanism.
Small molecule FGFR inhibitors
Several small-molecule FGFR inhibitors have been developed to treat cancers with FGFR alterations, such as urothelial cancer, breast cancer, gastric cancer, liver cancer, lung cancer, ovarian cancer, and cervical cancer. These inhibitors can be broadly classified into two categories based on their ability to bind to the ATP binding site of the tyrosine kinase domain: multikinase FGFR inhibitors and selective FGFR inhibitors.
Multikinase FGFR inhibitors, including derazatinib, dovitinib, tasurgratinib, lenvatinib, lucitanib, nintedanib, and ponatinib, exhibit activity against a variety of receptor tyrosine kinases (RTKs) in addition to FGFR. However, their broad activity leads to complex mechanisms of action and adverse effects compared to selective FGFR inhibitors.
Selective FGFR inhibitors include pan-FGFR inhibitors, FGFR1/2/3 inhibitors, and selective inhibitors for FGFR2, FGFR3, or FGFR4. Examples of pan-FGFR inhibitors are erdafitinib, futibatinib, rogaratinib, and resigratinib; FGFR1/2/3 inhibitors include pemigatinib, infigratinib, fexagratinib, and zoligratinib; Lirafugratinib is a selective FGFR2 inhibitor, and LOXO-435 is a selective FGFR3 inhibitor. Roblitinib and fissotioninib are selective FGFR4 inhibitors.
The U.S. Food and Drug Administration (FDA) has accelerated the approval of erdafitinib, futibatinib, infigratinib, and pemigatinib in second-line or later-line treatment for these indications based on the efficacy and tolerability observed in phase II trials. For example, in the clinical study of erdafitinib (BLC2001) for locally advanced and/or unresectable urothelial carcinoma with FGFR alterations, the overall response rate (ORR) was 40%, with a median progression-free survival (PFS) of 5.5 months and median overall survival (mOS) of 13.8 months.
In the phase II study of FIGHT-203, pemigatinib showed promising results in patients with myeloid and lymphoid neoplasms with FGFR1 fusions, with a 77% complete response rate. Adverse events such as anemia, stomatitis, and extremity pain were reported, but the drug was approved for relapsed and/or refractory myeloid and lymphoid neoplasms carrying FGFR1 fusions.
Hyperphosphatemia is a common adverse event with pan-FGFR inhibitors or FGFR1/2/3 inhibitors, likely due to inhibition of FGF23 signaling, which regulates phosphate absorption in the kidney. The development of FGFR2-specific and FGFR3-specific inhibitors aims to reduce this adverse reaction.
FGFR inhibitory monoclonal antibodies
Among biologics targeting FGF/FGFR signaling, bemarituzumab stands out as the sole candidate currently undergoing Phase III trials. It is a humanized anti-FGFR2b antibody designed to inhibit the binding of FGF7, FGF10, and FGF22, thereby blocking ligand-induced FGFR2b signaling in cancer cells. Additionally, due to its modified Fc domain, bemarituzumab can inhibit the human FcγRIIIA receptor on natural killer cells, enhancing its affinity to the body and enabling it to induce antibody-dependent cell-mediated cytotoxicity (ADCC).
In a Phase I trial involving patients with advanced FGFR2b-overexpressing gastric and gastroesophageal junction adenocarcinoma (GEA), bemarituzumab monotherapy exhibited acceptable tolerability, with an objective response rate (ORR) of 18% (5/28). Subsequently, bemarituzumab was investigated in combination with mFOLFOX6 (fluoropyrimidine, leucovorin, and oxaliplatin) in the Phase II FIGHT trial among patients with GEA harboring FGFR2 amplification or FGFR2 overexpression. The ORR among patients receiving bemarituzumab combined with chemotherapy was 53%, compared to 40% among those receiving placebo combined with chemotherapy. The median progression-free survival (PFS) was 9.5 months in the bemarituzumab group and 7.4 months in the placebo group (P=0.073). Several other trials are ongoing to evaluate bemarituzumab in combination with mFOLFOX6 plus (NCT0511626) or without (NCT050252801) nivolumab in patients with previously untreated advanced GEA.
Novel FGFR-targeted therapy
Proteolysis-targeting chimeras (PROTACs), chimeric antigen receptor (CAR) T cells, antibody-drug conjugates (ADCs), radioimmunoconjugates (RICs), and soluble receptors are innovative strategies for targeting the FGF-FGFR signaling pathway. These modalities hold promise as potential new treatments and have either been tested or are currently undergoing clinical trials.
PROTAC
PROTACs possess a dual moiety that allows them to interact with both target proteins and E3 ubiquitin ligases, leading to the ubiquitination-mediated proteasomal degradation of the target proteins. An example of this is LC-MB12, an infigratinib-based PROTAC that selectively degrades FGFR2, inhibiting the proliferation of Ba/F3 cells expressing TEL–FGFR2 fusion and FGFR2-amplified SNU16 cancer cells. In contrast, DGY-09-192 is another infigratinib-based PROTAC designed to recruit von Hippel–Lindau proteins (VHL), preferentially degrading FGFR1 and FGFR2. These drugs are currently in preclinical development.
CAR-T cells
In pediatric rhabdomyosarcoma, FGFR4 is frequently overexpressed, primarily due to its downstream effects on PAX3–FOXO1. However, before treatment, resistance mutations like N535K and V550L often emerge, potentially reducing the efficacy of small molecule inhibitors. To address this, researchers have created FGFR4-targeting CAR-T cells, such as RJ154-HL and 3A11, which feature distinct FGFR4-targeting single-chain variable fragments (scFvs). Currently, CAR-T cells designed to target FGFR are still in the preclinical development phase.
Antibody-based drugs
Aprutumab is a fully human anti-FGFR2 antibody that recognizes epitopes around P23, L27, and E29 in the common N-terminal regions of FGFR2b and FGFR2c isoforms. It has been shown to induce the internalization and trafficking of these variants to lysosomes for degradation. Both aprutumab and bemarituzumab demonstrated good safety and tolerability in phase I clinical trials. Currently, bemarituzumab is undergoing phase II and phase III clinical trials as a combination therapy, while aprutumab is being utilized in the development of antibody-drug conjugates (ADCs) and radioimmunoconjugates (RICs).
Aprutumab ixadotin (formerly BAY-1187982) is an anti-FGFR2 ADC with a non-cleavable linker, while LY3076226 is an anti-FGFR3 ADC with a cleavable linker. However, a phase 1 trial (NCT022368951) testing both drugs was terminated due to poor tolerability and limited activity.
Additionally, aprutumab and the anti-FGFR3 antibody vofatamab are employed for FGFR2-targeted RIC (227Th-Aprutumab, BAY 2304058) and FGFR3-targeted RIC (225Ac-vofatamab, FPI-1966 and 111In vofatamab, FPI-1967), respectively. BAY 2304058 remains in preclinical development, while FPI-1966 (for radioimmunotherapy) and FPI-1967 (for radiography) are currently being evaluated in a Phase I/II trial (NCT05363605) involving patients with advanced solid tumors expressing FGFR3.
Soluble FGFR receptor
Soluble FGFR receptors, like FP-1039 derived from the extracellular domain of FGFR1c, and Recifecept derived from the extracellular domain of FGFR3c, are recombinant protein drugs that act as decoy receptors, capturing FGFR ligands. FP-1039 exhibits a high affinity for mitogenic FGF ligands and has demonstrated antitumor effects in mouse xenograft models of FGFR1-amplified lung cancer and FGFR2-mutated endometrial cancer. The Phase 1 trial of FP-1039 indicated acceptable tolerability. A Phase Ib trial testing FP-1039 in combination with various chemotherapy regimens (NCT01868022) showed an objective response rate (ORR) of 47% in patients with squamous non-small cell lung cancer who received paclitaxel and carboplatin concurrently, and an ORR of 39% in patients with malignant pleural mesothelioma treated with pemetrexed and cisplatin.
Currently, small-molecule FGFR inhibitors and FGFR-targeting antibody drugs have been developed and tested in late-stage clinical trials. Several small-molecule FGFR inhibitors have been approved for use in cancer patients with FGFR alterations. However, certain adverse events, such as hyperphosphatemia and acquired resistance due to off-target inhibition of FGFR1, still limit the clinical benefit of these drugs. Selective third-generation inhibitors, particularly those targeting FGFR2 or FGFR3 specifically, are in clinical development. These drugs may avoid or significantly reduce the risk of hyperphosphatemia and could potentially overcome mutation-mediated resistance.
Conclusion
In conclusion, research on fibroblast growth factor receptors (FGFRs) has significantly advanced our understanding of their role in cancer and developmental disorders. Dysregulation of FGFR signaling is implicated in various cancers, making FGFRs attractive targets for therapy. The development of FGFR inhibitors, such as erdafitinib and pemigatinib, has shown promising results in clinical trials for treating FGFR-altered cancers. Additionally, ongoing research continues to elucidate the mechanisms of FGFR signaling, providing insights into potential therapeutic strategies for FGFR-related disorders. Overall, the progress in FGFR research highlights the potential of targeted therapies in cancer treatment and underscores the importance of continued research in this field.
Reference
- Katoh M. Fibroblast growth factor receptors as treatment targets in clinical oncology. Nat Rev Clin Oncol. 2019 Feb;16(2):105-122. doi: 10.1038/s41571-018-0115-y. PMID: 30367139.
- Bansal P, Dwivedi DK, Hatwal D, Sharma P, Gupta V, Goyal S, Maithani M. Erdafitinib as a Novel and Advanced Treatment Strategy of Metastatic Urothelial Carcinoma. Anticancer Agents Med Chem. 2021;21(18):2478-2486. doi: 10.2174/1871520621666210121093852. PMID: 33475078.
- Bansal P, Dwivedi DK, Hatwal D, Sharma P, Gupta V, Goyal S, Maithani M. Erdafitinib as a Novel and Advanced Treatment Strategy of Metastatic Urothelial Carcinoma. Anticancer Agents Med Chem. 2021;21(18):2478-2486. doi: 10.2174/1871520621666210121093852. PMID: 33475078.
- Javle M, Roychowdhury S, Kelley RK, Sadeghi S, Macarulla T, Weiss KH, Waldschmidt DT, Goyal L, Borbath I, El-Khoueiry A, Borad MJ, Yong WP, Philip PA, Bitzer M, Tanasanvimon S, Li A, Pande A, Soifer HS, Shepherd SP, Moran S, Zhu AX, Bekaii-Saab TS, Abou-Alfa GK. Infigratinib (BGJ398) in previously treated patients with advanced or metastatic cholangiocarcinoma with FGFR2 fusions or rearrangements: mature results from a multicentre, open-label, single-arm, phase 2 study. Lancet Gastroenterol Hepatol. 2021 Oct;6(10):803-815. doi: 10.1016/S2468-1253(21)00196-5. Epub 2021 Aug 3. PMID: 34358484.
- Goyal L, Meric-Bernstam F, Hollebecque A, Valle JW, Morizane C, Karasic TB, Abrams TA, Furuse J, Kelley RK, Cassier PA, Klümpen HJ, Chang HM, Chen LT, Tabernero J, Oh DY, Mahipal A, Moehler M, Mitchell EP, Komatsu Y, Masuda K, Ahn D, Epstein RS, Halim AB, Fu Y, Salimi T, Wacheck V, He Y, Liu M, Benhadji KA, Bridgewater JA; FOENIX-CCA2 Study Investigators. Futibatinib for FGFR2-Rearranged Intrahepatic Cholangiocarcinoma. N Engl J Med. 2023 Jan 19;388(3):228-239. doi: 10.1056/NEJMoa2206834. PMID: 36652354.
- Katoh M, Loriot Y, Brandi G, Tavolari S, Wainberg ZA, Katoh M. FGFR-targeted therapeutics: clinical activity, mechanisms of resistance and new directions. Nat Rev Clin Oncol. 2024 Feb 29. doi: 10.1038/s41571-024-00869-z. Epub ahead of print. PMID: 38424198.