Allosteric Inhibitors for EGFR-Driven Cancers: An Alternative Therapeutic Approach
Abstract
EAI045 and JBJ-04-125-02 are two allosteric inhibitors that have been studied extensively as potential alternative therapeutic options for EGFR-targeted cancer treatments. These inhibitors work by binding to a specific site on the EGFR protein, which causes a conformational change that prevents the protein from being activated and signaling for cell growth and division.
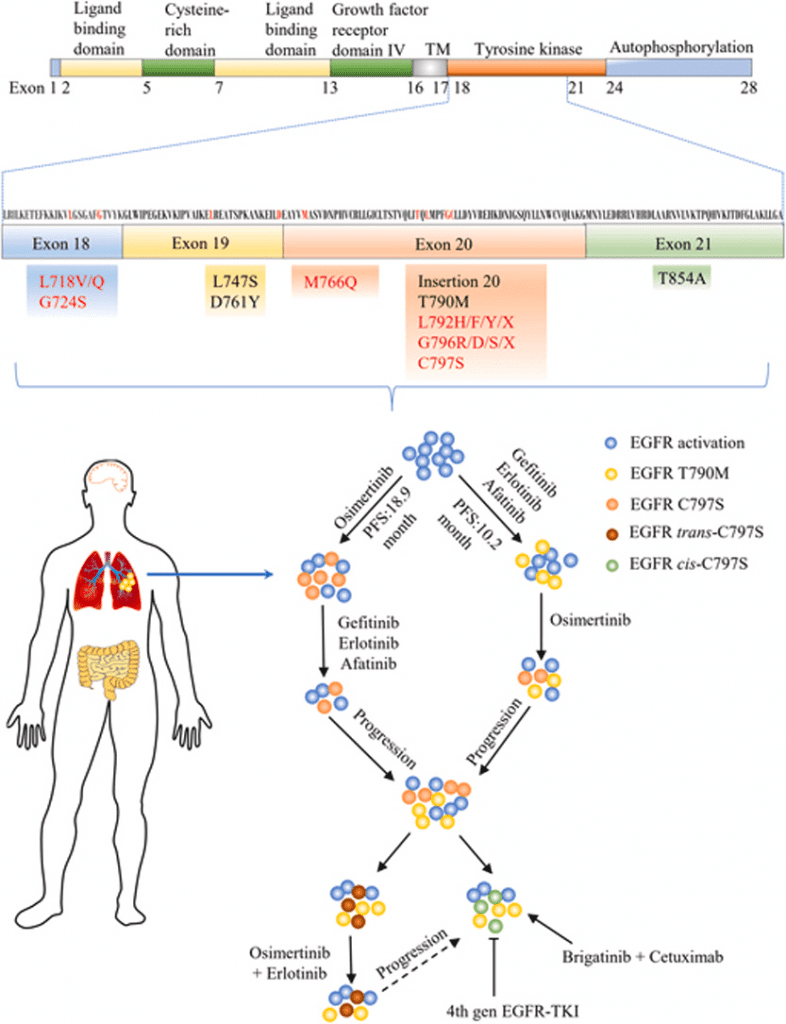
Fig 1. The clinical progress and promising directions for novel drug development of overcoming EGFR TKIs resistance.[1]
EGFR and its role in cancer
EGFR, also known as ErbB1, is a transmembrane receptor protein that plays a critical role in the development and progression of cancer. EGFR is involved in several signaling pathways that regulate cell growth, proliferation, differentiation, and survival. Aberrant activation of the EGFR pathway is a hallmark of many types of cancer, including lung, breast, and colorectal cancer.
EGFR is frequently overexpressed or mutated in cancer cells, leading to constitutive activation of the EGFR signaling pathway. This activation can drive uncontrolled cell growth, invasion, and metastasis, promoting tumor progression and resistance to chemotherapy and radiation therapy.
Given its central role in cancer progression, EGFR has become an important therapeutic target in oncology. Several EGFR-targeted therapies have been developed, including monoclonal antibodies that bind to the extracellular domain of EGFR and small molecule inhibitors that target the ATP-binding site in the intracellular domain of EGFR. These therapies have shown significant clinical benefits in some patients, particularly in those with EGFR-activating mutations or overexpression.
The limitations of current EGFR-targeted therapies
Despite the significant progress made in the development of EGFR-targeted therapies, these therapies still have several limitations that need to be addressed. Firstly, these therapies only work in patients whose tumors harbor EGFR-activating mutations or overexpression, which represent a minority of patients. Therefore, the identification of biomarkers that can accurately predict which patients will benefit from EGFR-targeted therapies is critical.
Secondly, the development of drug resistance is a major challenge in EGFR-targeted therapy. Resistance can arise through various mechanisms, such as secondary mutations in the EGFR kinase domain, activation of alternative signaling pathways, and alterations in downstream effectors. Developing strategies to overcome or prevent drug resistance is crucial for improving the efficacy of EGFR-targeted therapies.
Finally, EGFR-targeted therapies are associated with a range of side effects, including skin rash, diarrhea, and pulmonary toxicity. These side effects can affect patients’ quality of life and can limit the duration of treatment. Therefore, the development of more specific and less toxic EGFR-targeted therapies is necessary.
Allosteric inhibitors as an alternative therapeutic approach
Allosteric inhibitors are a class of small molecule compounds that bind to a specific site on a protein molecule other than its active site. This site, known as the allosteric site, is located away from the enzyme’s active site and can regulate the protein’s function by changing its conformation. Allosteric inhibitors can modulate protein activity in several ways, such as inhibiting or enhancing its activity, stabilizing or destabilizing its conformation, or disrupting protein-protein interactions.
Allosteric inhibitors represent a promising alternative therapeutic approach to traditional inhibitors that target the active site. One of the advantages of allosteric inhibitors is that they can potentially bind to more specific and less conserved sites on the protein, reducing the likelihood of off-target effects and minimizing toxicity. Additionally, allosteric inhibitors can overcome drug resistance, as they can bind to alternative sites on the protein and inhibit its activity through a different mechanism.
Several allosteric inhibitors have been developed for the treatment of various diseases, including cancer. For instance, allosteric inhibitors targeting the EGFR receptor have shown promising results in preclinical studies, demonstrating improved potency and selectivity compared to traditional inhibitors. Combination therapy with allosteric inhibitors and traditional inhibitors has also shown synergistic effects, suggesting that these therapies may have increased efficacy when used in combination.
EAI045 has been shown to be highly effective against EGFR mutant lung cancers and has also demonstrated activity against other types of cancer that rely on EGFR signaling. In preclinical studies, EAI045 has been shown to have minimal toxicity and excellent pharmacokinetic properties, making it a promising candidate for further development.
Similarly, JBJ-04-125-02 has shown potent activity against EGFR-driven cancers in preclinical studies. It has been shown to have good oral bioavailability and excellent pharmacokinetics, indicating that it has the potential to be an effective treatment option for patients.
While these two inhibitors have shown promise in preclinical studies, further research is needed to determine their safety and efficacy in humans. Clinical trials will be necessary to determine the optimal dosing and administration strategies for these inhibitors and to evaluate their long-term safety and efficacy in patients.
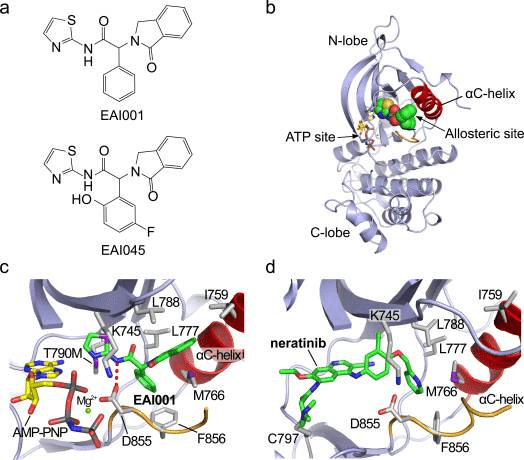
Fig 2. Structure and binding mode of allosteric EGFR inhibitors [4]
a. Chemical structures of EAI001 and EAI045. b. Overall view of the structure of T790M/V948R EGFR bound to EAI001 and AMP-PNP. EAI001 is shown in CPK form with carbon atoms in green. The V948R substitution was introduced to allow crystallization of the kinase in an inactive conformation8. c. Detailed view of the interactions of EAI001. A hydrogen bond with Asp855 in the “DFG” segment of the kinase is shown as a dashed red line. d. The structure of irreversible inhibitor neratinib bound to EGFR T790M (PDB ID 2JIV). Neratinib occupies the ATP site, but also extends into the allosteric pocket occupied by EAI001.
EAI045 is a first EGFR allosteric inhibitor
EAI045 is a small molecule allosteric inhibitor that targets activating mutations in the epidermal growth factor receptor (EGFR) in cancer cells. It has been shown to have promising in vitro and in vivo activity against various EGFR-mutant cancer cell lines, including those that are resistant to first- and second-generation EGFR inhibitors.
In vitro, EAI045 was found to be more potent than first-generation EGFR inhibitors such as erlotinib and gefitinib in inhibiting cell proliferation of EGFR-mutant cell lines. Additionally, EAI045 was able to inhibit the phosphorylation of EGFR in these cells, which is an important driver of cancer growth.
In vivo, EAI045 was shown to be effective in inhibiting tumor growth in mouse xenograft models of EGFR-mutant lung cancer. Importantly, EAI045 was also able to inhibit the growth of EGFR-mutant cancer cells that had become resistant to first- and second-generation EGFR inhibitors, suggesting that it may be a promising therapeutic option for patients who have developed resistance to these drugs.
Interestingly, EAI045 has been shown to have a high degree of selectivity for EGFR mutants over the wild-type EGFR, which is expressed in normal cells. This selectivity is thought to be due to the unique structural features of the mutant EGFR, which create a specific binding pocket for the inhibitor.
Overall, the promising in vitro and in vivo activity data for EAI045 suggest that it may be a valuable therapeutic option for patients with EGFR-mutant cancers, including those who have developed resistance to first- and second-generation EGFR inhibitors. However, further clinical studies are needed to fully evaluate its safety and efficacy in human patients.
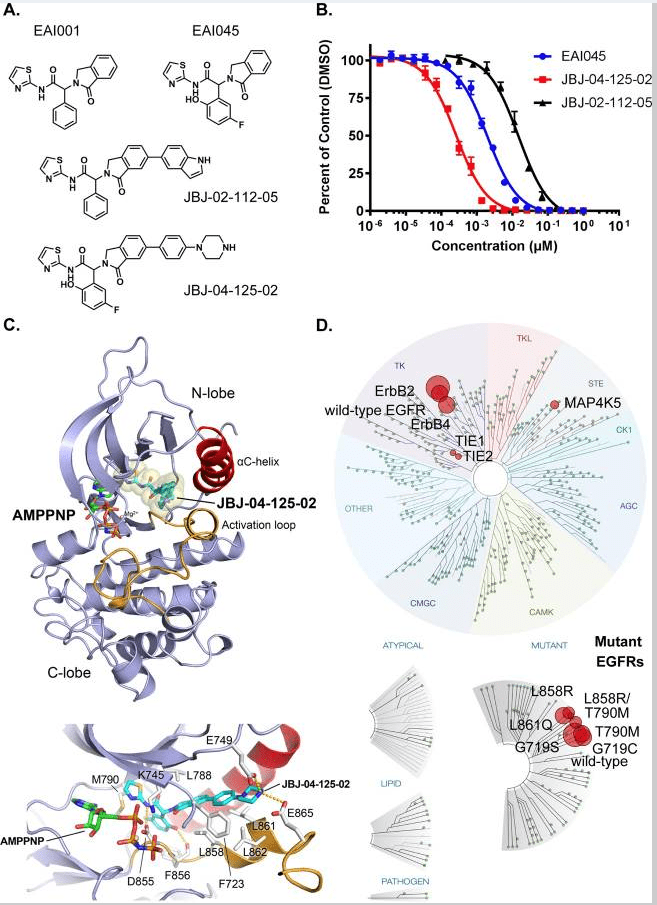
Fig 3. Structural properties and biochemical analyses of the allosteric inhibitor, JBJ-04-125-02[5].
A. Molecular structures of EAI045, EAI001, JBJ-04-125-02, and JBJ-02-112-05. B. In vitro inhibition of EGFR L858R/T790M kinase by allosteric inhibitors. Enzyme activity was measured using an HTRF-based assay in the presence of increasing concentrations of each inhibitor as indicated. Fractional activity is relative to a 1% DMSO control. C. Crystal structure of EGFR (T790M/V948R) bound to JBJ-04-125-02 and AMP-PNP. JBJ-04-125-02 is displayed using CPK-coloring with cyan carbon atoms. Distinct hydrogen bonds are shown as a dashed line. D. Kinome selectivity obtained from KINOMEscan (DiscoverX) using 10μM of JBJ-04-125-02 against 468 kinases. The size of circles mapped onto the kinase phylogenetic tree utilizing DiscoverX TREEspot™ corresponds to the strength of the binding affinity as indicated in the figure. S-score (35) indicates the relative selectivity of the compound with 35% cut-off (number of non-mutant kinases with <35% control/number of non-mutant kinases tested).
JBJ-04-125-02 as an allosteric inhibitor that can target both wild-type and mutant forms of EGFR
The development of allosteric inhibitors has shown great potential as an alternative therapeutic approach for the treatment of cancer. Recently, an allosteric inhibitor known as JBJ-04-125-02 has been developed that target both the wild-type and mutant forms of EGFR, a critical receptor tyrosine kinase involved in cancer progression.
JBJ-04-125-02 was developed by a research team using a structure-based design approach. The inhibitor selectively targets the EGFR kinase domain allosteric site, leading to the inhibition of EGFR activity through a distinct mechanism from that of traditional inhibitors that target the ATP-binding site. This approach overcomes the limitations of traditional inhibitors, which can lead to drug resistance due to mutations in the ATP-binding site.
In vitro studies have shown that JBJ-04-125-02 is highly effective in inhibiting the activity of wild-type EGFR and several EGFR mutants, including the T790M and L858R mutations, which are common in non-small cell lung cancer (NSCLC) patients. Furthermore, JBJ-04-125-02 showed significantly lower toxicity to normal cells than traditional inhibitors that target the ATP-binding site.
In vivo studies using mouse models of NSCLC have shown that JBJ-04-125-02 inhibits tumor growth and metastasis, suggesting that this inhibitor could be a promising therapeutic agent for the treatment of NSCLC. Interestingly, JBJ-04-125-02 has also been shown to inhibit the activity of the EGFR variant III (EGFRvIII), which is expressed in glioblastoma, an aggressive brain tumor.
In summary, the development of JBJ-04-125-02, an allosteric inhibitor that targets both wild-type and mutant forms of EGFR, represents a significant advancement in the development of therapeutic agents for the treatment of cancer. This inhibitor overcomes the limitations of traditional inhibitors, which can lead to drug resistance due to mutations in the ATP-binding site. The potential of JBJ-04-125-02 as a therapeutic agent for the treatment of NSCLC and other EGFR-driven cancers warrants further investigation.
The molecular basis for the cooperative binding and synergy of ATP-site and allosteric EGFR inhibitors
The molecular basis for the cooperative binding and synergy of ATP-site and allosteric EGFR inhibitors has been studied to better understand the mechanism of action of these inhibitors and their potential for use in cancer treatment. A recent study has shed light on the molecular basis for the cooperative binding and synergy of these inhibitors.
The study showed that the binding of allosteric inhibitors to EGFR induces conformational changes in the protein, leading to the formation of a hydrophobic pocket that is adjacent to the ATP-binding site. This pocket creates a site where ATP-site inhibitors can bind, leading to cooperative binding and synergy between the two types of inhibitors.
Furthermore, the study demonstrated that this cooperative binding and synergy is dependent on the presence of specific amino acid residues in the EGFR protein. Mutations in these residues can affect the ability of the two types of inhibitors to bind and cooperate with each other.
These findings suggest that the cooperative binding and synergy of ATP-site and allosteric inhibitors is a result of the conformational changes induced by the binding of the allosteric inhibitor to EGFR. This mechanism provides a potential strategy for developing more effective EGFR inhibitors for the treatment of cancer.
In conclusion, the molecular basis for the cooperative binding and synergy of ATP-site and allosteric EGFR inhibitors has been elucidated, providing insights into the mechanism of action of these inhibitors and their potential for use in cancer treatment. The identification of specific amino acid residues that are critical for this mechanism provides a potential strategy for developing more effective EGFR inhibitors for the treatment of cancer.
Allosteric inhibitors as a promising alternative for EGFR-targeted therapies
EGFR (Epidermal Growth Factor Receptor) is a protein that is involved in the growth and proliferation of many types of cancer cells. EGFR-targeted therapies, such as tyrosine kinase inhibitors (TKIs) and monoclonal antibodies, have been successful in treating various cancers by blocking the activation of EGFR. However, the emergence of drug resistance and adverse effects have led to the search for alternative therapeutic approaches.
One such approach is the use of allosteric inhibitors, which target a different site on the EGFR protein than the active site targeted by TKIs and antibodies. Allosteric inhibitors bind to a specific site on the EGFR protein, causing a conformational change that prevents the protein from being activated. This approach offers several advantages over traditional EGFR-targeted therapies. For instance, allosteric inhibitors have been shown to be effective against certain EGFR mutations that are resistant to TKIs, potentially expanding the range of cancers that can be treated.
Another advantage of allosteric inhibitors is that they may have fewer side effects than traditional EGFR-targeted therapies. This is because allosteric inhibitors target a different site on the protein and may have a more specific and selective mode of action, reducing the risk of off-target effects. Additionally, because allosteric inhibitors do not directly inhibit the active site of EGFR, they may be less likely to induce drug resistance.
Despite the promising potential of allosteric inhibitors as an alternative therapeutic approach for EGFR-targeted therapies, further research is needed to optimize their effectiveness and safety. For example, the design and optimization of allosteric inhibitors is challenging due to the complexity of the EGFR protein and the diversity of EGFR mutations found in different cancers. Therefore, continued research is needed to identify new allosteric sites on EGFR and to develop allosteric inhibitors that are effective against a broad range of EGFR mutations.
In conclusion, allosteric inhibitors offer a promising alternative therapeutic approach for EGFR-targeted therapies in the treatment of various types of cancer. The development of allosteric inhibitors could potentially overcome drug resistance and reduce side effects associated with traditional EGFR-targeted therapies. However, further research is necessary to optimize the efficacy and safety of allosteric inhibitors, and to identify new allosteric sites on the EGFR protein. This will help to expand the range of cancers that can be treated with allosteric inhibitors and improve cancer treatment options for patients.
Reference
- Dong, R. F., Zhu, M. L., Liu, M. M., Xu, Y. T., Yuan, L. L., Bian, J., … & Kong, L. Y. (2021). EGFR mutation mediates resistance to EGFR tyrosine kinase inhibitors in NSCLC: From molecular mechanisms to clinical research. Pharmacological research, 167, 105583.
- To, C., Beyett, T. S., Jang, J., Feng, W. W., Bahcall, M., Haikala, H. M., … & Jänne, P. A. (2022). An allosteric inhibitor against the therapy-resistant mutant forms of EGFR in non-small cell lung cancer. Nature Cancer, 3(4), 402-417.
- Beyett, T. S., To, C., Heppner, D. E., Rana, J. K., Schmoker, A. M., Jang, J., … & Eck, M. J. (2022). Molecular basis for cooperative binding and synergy of ATP-site and allosteric EGFR inhibitors. Nature Communications, 13(1), 2530.
- Jia, Y., Yun, C. H., Park, E., Ercan, D., Manuia, M., Juarez, J., … & Eck, M. J. (2016). Overcoming EGFR (T790M) and EGFR (C797S) resistance with mutant-selective allosteric inhibitors. Nature, 534(7605), 129-132.
- To, C., Jang, J., Chen, T., Park, E., Mushajiang, M., De Clercq, D. J., … & Jänne, P. A. (2019). Single and Dual Targeting of Mutant EGFR with an Allosteric InhibitorSingle-agent Allosteric EGFR Inhibitor. Cancer discovery, 9(7), 926-943.
- Qiu, Y., Yin, X., Li, X., Wang, Y., Fu, Q., Huang, R., & Lu, S. (2021). Untangling dual-targeting therapeutic mechanism of epidermal growth factor receptor (EGFR) based on reversed allosteric communication. Pharmaceutics, 13(5), 747.