Comparative Overview of Tyvaso Nebulizer and Dry Powder Inhaler (DPI) for Pulmonary Hypertension Treatment
Abstract
Pulmonary Arterial Hypertension (PAH) and Pulmonary Hypertension due to Interstitial Lung Disease (PH-ILD) are severe, progressive conditions characterized by elevated pressure in the pulmonary arteries. PAH is primarily driven by endothelial dysfunction, smooth muscle proliferation, and vascular remodeling, while PH-ILD involves additional mechanisms such as lung parenchymal destruction and chronic hypoxia. Treprostinil, a synthetic prostacyclin analog, plays a critical role in managing these diseases by inducing vasodilation and preventing further vascular remodeling. The inhaled formulation of treprostinil, particularly the dry powder inhaler (Tyvaso DPI), has proven to be highly effective, improving exercise capacity and reducing symptoms in both PAH and PH-ILD. The INCREASE trial demonstrated the efficacy of inhaled treprostinil in PH-ILD patients, marking a significant advancement in treatment options for this condition. Overall, treprostinil remains a cornerstone therapy for managing PAH and PH-ILD, offering patients better outcomes and improved quality of life.
Introduction
Pulmonary hypertension (PH) is a common disease whose core functions is elevated pressure in the pulmonary arteries. Though variety of tools are used to evaluate and monitor the disease, the gold standard for diagnosis of PH remains right heart catheterization. The thresholds for making the diagnosis of PH were recently changed by the 6th World Symposium on pulmonary Hypertension. The professionals revised the criteria for precapillary PH to a mean pulmonary arterial pressure (mPAP) of >20 mmHg with a pulmonary vascular resistance of 2 Wood units or greater.
The management and evaluation of PH depend largely on identifying the underlying cause of increased pulmonary pressures. PH is classified into five groups according to the World Health Organization (WHO):
- Group I: Pulmonary arterial hypertension (PAH)
- Group II: PH related to left heart disease
- Group III: PH related to chronic lung disease
- Group IV: PH due to pulmonary obstructions, such as chronic thromboembolic pulmonary hypertension (CTEPH)
- Group V: PH due to multifactorial mechanisms
In this context, we focus on PAH (Group I) and PH related to interstitial lung disease (PH-ILD, Group III), where prostacyclin therapies have shown significant therapeutic benefits.
PAH is a progressive and complex disease that remains incurable. It is characterized by increasing pulmonary vascular resistance, which leads to right ventricular failure and, if untreated, death. Environmental and genetic factors contribute to endothelial and smooth muscle cell proliferation in the pulmonary arteries, causing medial hypertrophy and intimal fibrosis. This process is driven by multiple overlapping pathways, including chronic inflammation, oxidative stress, metabolic dysfunction, and resistance to cell death. To cope with the high resistance, the right ventricle undergoes hypertrophy to maintain cardiac output, but this adaptation eventually becomes maladaptive, resulting in right ventricular dilation, tricuspid regurgitation, and dysfunction.
While the pathophysiological mechanisms of PH due to interstitial lung disease share some similarities with PAH, they remain distinct and not fully understood. The destruction of lung parenchyma and vasculature through fibrosis and chronic hypoxia is now believed to be accompanied by endothelial changes and remodeling, similar to PAH. Genetic and molecular factors also play a significant role in these vascular changes, occurring somewhat independently of the underlying lung disease. Further research is needed to identify which patients with lung disease may benefit from pulmonary vasodilator therapy, as only one therapy has been proven effective in treating PH-ILD.
For PAH, pulmonary vasodilators are the mainstay of treatment. Calcium channel blockers, once a cornerstone of PAH management, are now only used in a limited subset of patients who show a positive hemodynamic response on right heart catheterization. Careful monitoring is required in such cases to prevent hemodynamic deterioration. In addition, riociguat is FDA-approved for patients with CTEPH (Group IV PH) who are not candidates for surgery or have persistent PH after surgery.
However, studies investigating pulmonary vasodilators for PH in groups outside of PAH and CTEPH have often been disappointing, with some showing adverse outcomes. Therefore, routine use of vasodilators is not recommended in these cases. Patients with severe symptoms or PH that is disproportionate to their underlying disease should be referred to specialized centers for advanced management. A major development for PH-ILD has been the introduction of inhaled treprostinil, supported by the results of the INCREASE trial, which has provided evidence for its efficacy in treating Group III PH.
Currently, FDA-approved therapies for PAH target three main pathways: nitric oxide, endothelin, and prostacyclin. The prostacyclin pathway, the oldest of these, offers the greatest variety in delivery methods, including subcutaneous, intravenous, oral, and inhaled formulations.
Overview of Pulmonary Arterial Hypertension (PAH)
Pulmonary Arterial Hypertension (PAH) is a severe, progressive disease characterized by elevated blood pressure in the pulmonary arteries, the vessels that carry blood from the heart to the lungs. PAH is classified as Group I by the World Health Organization (WHO) within the broader spectrum of pulmonary hypertension disorders. This condition occurs when the small arteries in the lungs narrow, thicken, or become blocked, increasing resistance to blood flow. As a result, the right side of the heart must work harder to pump blood through these arteries, which can lead to right ventricular hypertrophy, heart failure, and eventually death if left untreated.
The exact causes of PAH can vary. It may be idiopathic, heritable, or associated with conditions such as connective tissue diseases, congenital heart defects, and certain drugs or toxins. Regardless of the underlying cause, the hallmark of PAH is a relentless increase in pulmonary vascular resistance. This heightened resistance is driven by several overlapping mechanisms, including endothelial dysfunction, chronic inflammation, and vascular remodeling, all of which contribute to the narrowing of the pulmonary arteries.
Despite its complexity, PAH has seen significant advances in treatment over the past few decades. Current therapies aim to reduce symptoms, improve quality of life, and slow disease progression. These therapies primarily target three key pathways involved in the pathogenesis of PAH: the nitric oxide pathway, endothelin pathway, and prostacyclin pathway. While there is no cure, treatments such as inhaled prostacyclin analogs, including treprostinil, have shown significant efficacy in managing the symptoms and improving outcomes in PAH patients.
Pathophysiology of PAH and Pulmonary Hypertension Due to Interstitial Lung Disease (PH-ILD)
Pulmonary Arterial Hypertension (PAH) is driven by complex, interrelated mechanisms involving the pulmonary arteries. The pathophysiology of PAH centers on endothelial dysfunction, which leads to the overgrowth of smooth muscle cells and fibroblasts, causing narrowing and remodeling of the pulmonary arteries. This process results in increased pulmonary vascular resistance, ultimately elevating the pressure in the pulmonary arteries. Factors such as chronic inflammation, oxidative stress, and imbalances in vasoactive mediators like nitric oxide, prostacyclin, and endothelin contribute to these pathological changes. If left untreated, the high pulmonary artery pressure places significant strain on the right side of the heart, eventually leading to right ventricular failure.
In Pulmonary Hypertension due to Interstitial Lung Disease (PH-ILD), the pathophysiology shares some overlap with PAH but involves additional mechanisms related to lung tissue damage. In PH-ILD, the destruction of lung parenchyma due to fibrosis and chronic hypoxic pulmonary vasoconstriction plays a central role. These changes trigger remodeling of the pulmonary vasculature similar to that seen in PAH. However, in PH-ILD, there is a stronger interplay between the underlying lung disease and the vascular changes, making it more complex and difficult to treat.
While the mechanisms of PH-ILD remain less well understood, recent research indicates that endothelial cell injury and chronic inflammation may promote vascular remodeling in a manner analogous to PAH. However, fibrosis and destruction of the lung tissue in ILD further complicate the disease, making it more difficult to manage than isolated PAH. Advances in pulmonary vasodilator therapies, such as inhaled prostacyclins, have shown promise in managing PH-ILD, but treatment remains a challenge due to the complexity of the underlying lung disease.
Treprostinil and Its Role in PAH Management
Treprostinil is a synthetic prostacyclin analog used to manage Pulmonary Arterial Hypertension (PAH). Prostacyclin is a naturally occurring substance in the body that plays a critical role in vascular homeostasis by inducing vasodilation, inhibiting platelet aggregation, and protecting against vascular remodeling. Treprostinil mimics these effects, making it a key therapy in PAH, where pulmonary artery constriction and remodeling are primary drivers of disease progression. Treprostinil is available in several formulations, including intravenous, subcutaneous, oral, and inhaled forms, each offering different benefits in terms of convenience, efficacy, and patient compliance.
The inhaled formulation of treprostinil is particularly significant due to its targeted delivery directly to the lungs, minimizing systemic side effects while maximizing the therapeutic impact on the pulmonary vasculature. Inhaled treprostinil has been shown to improve exercise capacity and reduce symptoms in patients with PAH, making it a critical component of a multimodal approach to PAH management. The development of a dry powder inhaler (Tyvaso DPI) further enhances patient convenience, allowing for easier administration compared to nebulized formulations.
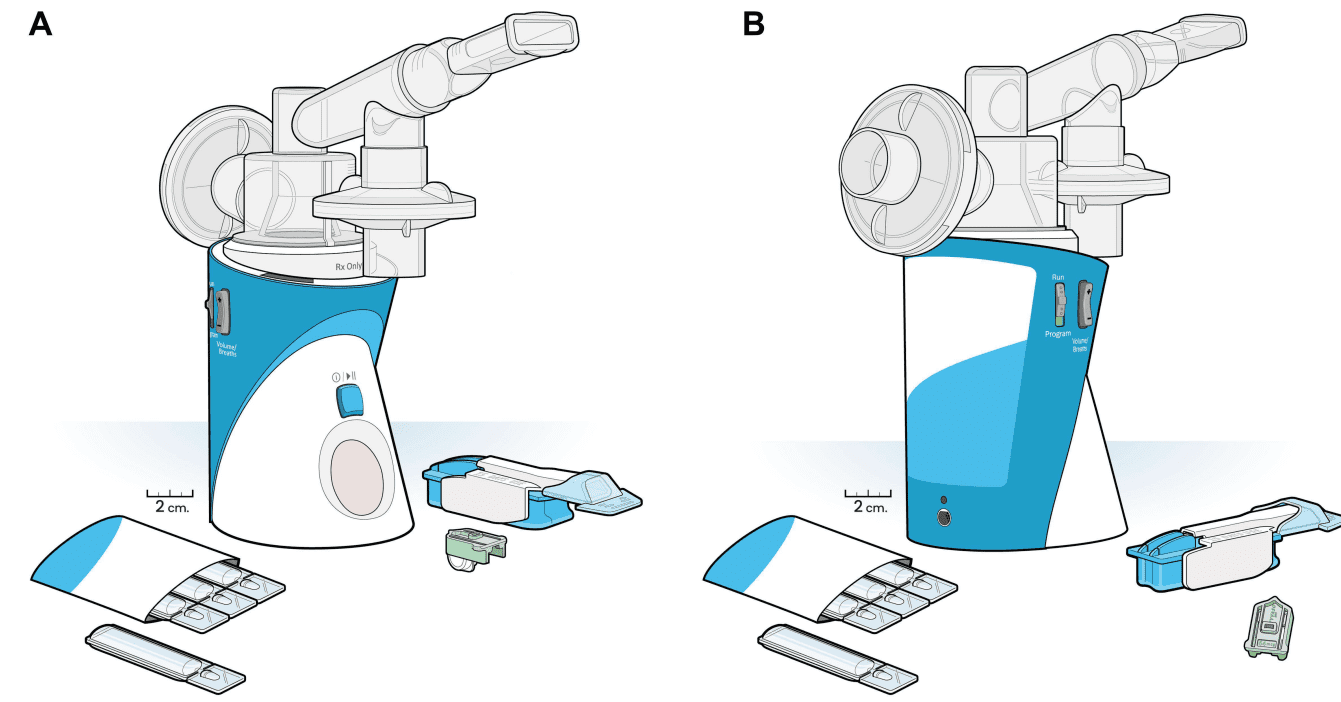
Figure 1 (A and B) Tyvaso delivery devices. Views of the Tyvaso nebulizer device and Tyvaso direct powder inhaler (DPI) device, with sizes to scale. Drug ampules (in envelope) are used with the nebulizer device, while cartridges (here, a light green 64 mcg dose cartridge) are used with the DPI device.
Clinical Evidence Supporting Inhaled Treprostinil
The INCREASE trial was a pivotal study that established the efficacy of inhaled treprostinil in patients with pulmonary hypertension due to interstitial lung disease (PH-ILD). In this randomized, controlled trial, patients treated with inhaled treprostinil showed significant improvements in exercise capacity, as measured by the six-minute walk distance (6MWD), compared to those receiving a placebo. Additionally, the study demonstrated reductions in clinical worsening and hospitalizations related to pulmonary hypertension.
The results of the INCREASE trial marked a breakthrough in the treatment of PH-ILD, a group for which few effective treatments existed. The trial also confirmed that inhaled treprostinil was generally well-tolerated, with manageable side effects, such as cough and headache. These findings highlight the importance of treprostinil in managing both PAH and PH-ILD, offering hope to patients with these progressive diseases.
References
- Cassady, S. J., Almario, J. A. N., & Ramani, G. V. (2024). Therapeutic potential of treprostinil inhalation powder for patients with pulmonary arterial hypertension: Evidence to date. Drug, Healthcare and Patient Safety, 16, 51-59.
- Galiè, N., Humbert, M., Vachiery, J. L., Gibbs, S., Lang, I., Torbicki, A., & Simonneau, G. (2016). 2015 ESC/ERS guidelines for the diagnosis and treatment of pulmonary hypertension. European Heart Journal, 37(1), 67-119.
- McLaughlin, V. V., Archer, S. L., Badesch, D. B., Barst, R. J., Farber, H. W., Lindner, J. R., & Rosenson, R. S. (2009). ACCF/AHA 2009 expert consensus document on pulmonary hypertension. Journal of the American College of Cardiology, 53(17), 1573-1619.
- Rubin, L. J. (1997). Primary pulmonary hypertension. New England Journal of Medicine, 336(2), 111-117.
- Thenappan, T., Ormiston, M. L., Ryan, J. J., & Archer, S. L. (2018). Pulmonary arterial hypertension: Pathogenesis and clinical management. BMJ, 360, j5492.
- Dorfmüller, P., Humbert, M., Perros, F., Sanchez, O., Simonneau, G., & Durand-Gasselin, I. (2003). Inflammation in pulmonary arterial hypertension. European Respiratory Journal, 22(2), 358-363.
- Stacher, E., Graham, B. B., Hunt, J. M., Gandjeva, A., Groshong, S. D., McLaughlin, V. V., … & Stacher-Priehse, E. (2012). Modern age pathology of pulmonary arterial hypertension. American Journal of Respiratory and Critical Care Medicine, 186(3), 261-272.
- Rabinovitch, M., Guignabert, C., Humbert, M., & Nicolls, M. R. (2014). Inflammation and immunity in the pathogenesis of pulmonary arterial hypertension. Circulation Research, 115(1), 165-175.
- Waxman, A., Restrepo-Jaramillo, R., Thenappan, T., & Hemnes, A. (2021). Inhaled treprostinil in pulmonary hypertension due to interstitial lung disease. New England Journal of Medicine, 384(4), 325-334.
- Gomberg-Maitland, M., Olschewski, H., & Farber, H. W. (2008). Prostacyclin therapies for the treatment of pulmonary arterial hypertension. European Respiratory Journal, 31(4), 891-901.
- King, C. S., Brown, A. W., & Shlobin, O. A. (2021). Treprostinil therapy for pulmonary hypertension in interstitial lung disease: a review of the INCREASE study. Respiratory Medicine, 187, 106575.