Histone deacetylase (HDAC) Inhibitors Overview
Abstract
Histone deacetylase (HDAC) is a protease that regulates gene expression by altering chromatin structure. HDACs remove acetyl groups from histones, causing chromatin to become more compact, which in turn reduces gene expression. In addition to gene regulation, HDACs are involved in a variety of cell physiological processes, such as cell division, differentiation, and apoptosis. Such complex physiological functions make HDAC a potential target for developing cancer therapies. In cancer cells, HDAC is usually overexpressed, which is conducive to the growth and survival of cancer cells. HDAC inhibitors work by inhibiting HDAC activity, causing changes in gene expression and cellular processes that inhibit cancer cell growth and lead to cancer cell death. There are various subtypes of HDACs, which have different functions and expressions in different cells, and may have potential therapeutic schemes for cancer and other diseases. As a result, there has been a significant increase in research related to HDAC in recent years.
The mechanism of action of HDAC in human body
HDACs, a group of enzymes, have a significant role in controlling gene expression in the human body. Their function involves eliminating acetyl groups from histone proteins, which play a crucial role in DNA packaging into chromatin. This activity leads to a denser chromatin structure that impedes the access of transcriptional machinery to DNA, resulting in reduced gene expression[1].
In addition to histones, HDACs also target other non-histone proteins, including transcription factors, cytoskeleton proteins, and chaperones[2]. By modifying the acetylation status of these proteins, HDACs[3] regulate diverse cellular processes, such as cell cycle progression, DNA damage repair, apoptosis, and differentiation(Figure 1).
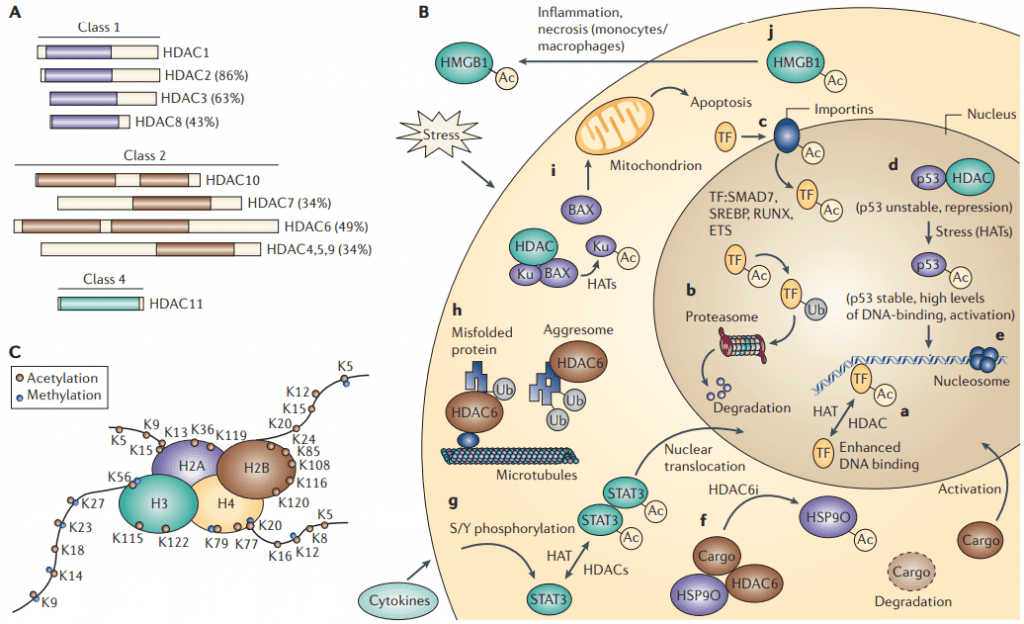
Figure 1 The acetylome. A | A schematic representation of histone deacetylases (HDACs) B | A schematic view of the functioning acetylome. C | The acetylation[3]
Four classes of HDACs exist in humans with varying structural and functional characteristics. Class I HDACs, such as HDAC1, 2, 3, and 8, are expressed ubiquitously and predominantly located in the nucleus. Class II HDACs, including HDAC4, 5, 6, 7, 9, and 10, are expressed in a tissue-specific manner and can move between the nucleus and cytoplasm. Class III HDACs, or sirtuins, require NAD+ as a cofactor, and include SIRT1-7. Class IV HDACs, like HDAC11, share structural features with both Class I and II HDACs and exhibit nuclear and cytoplasmic localization[4].
The dysregulation of HDAC activity has been implicated in the development and progression of various diseases, including cancer, neurodegeneration, and metabolic disorders. In particular, the aberrant overexpression and activity of HDACs have been found in many types of cancer, which has led to the development of HDAC inhibitors as a promising therapeutic strategy for cancer treatment[5].
HDAC inhibitors
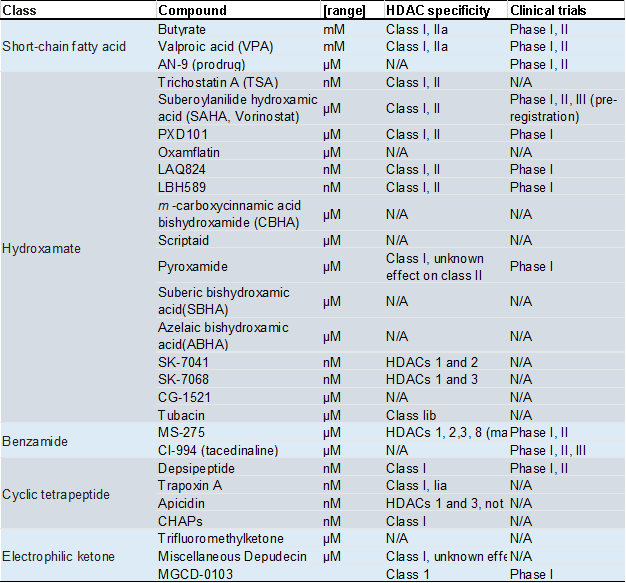
HDAC inhibitors (HDACi) are a class of small molecules that can selectively or non-selectively inhibit the activity of HDACs. By blocking the deacetylation of histones and non-histone proteins, HDACi can modulate gene expression and cellular processes in cancer cells, leading to cell cycle arrest, differentiation, and apoptosis. Bolden’s team published a review in 2006 that summarized the state of research on different types of HDACis at the time (Table 1) [5].
Table 1 Molecular characteristics and clinical trial status of HDACi
The molecular mechanism of HDACi action has been extensively studied, and several hypotheses have been proposed. One of the most widely accepted models is the “histone code” hypothesis, which suggests that the acetylation status of histones serves as a molecular code that governs gene expression[6]. According to this model, the acetylation of histones at specific lysine residues (e.g., H3K9 and H3K14) creates a relaxed chromatin structure that facilitates transcriptional activation, while the deacetylation of histones promotes a condensed chromatin structure and transcriptional repression.
Inhibiting HDAC activity using HDACi can disturb the histone code and result in the hyperacetylation of histones, which leads to a more relaxed chromatin structure. This effect can upregulate genes that induce cell cycle arrest, differentiation, and apoptosis while downregulating genes that promote cell survival and proliferation [5]. Additionally, disrupting protein-protein interactions is another proposed mechanism of HDACi action. HDACs interact with other proteins, such as transcription factors, co-repressors, and chaperones, through their deacetylase activity. When HDAC activity is inhibited by HDACi, these interactions can be disrupted, resulting in changes in cellular processes and gene expression.
Histone deacetylase inhibitors can trigger the DNA damage response (DDR) pathway and cause DNA damage. The DDR pathway is a signaling network that detects DNA damage and initiates various responses, such as cell cycle arrest, apoptosis, and DNA repair[7]. HDACi can disrupt chromatin structure, induce oxidative stress, and generate reactive oxygen species (ROS), leading to DNA damage. Activation of the DDR pathway by HDACi results in cell cycle arrest, apoptosis, and increased expression of DNA repair genes.
There is evidence indicating that HDACi can modulate the immune response by regulating the acetylation status of non-histone proteins in immune cells. An instance of this is HDACi’s capacity to boost the acetylation of STAT proteins, which partake in cytokine signaling and immune cell activation. This effect can result in the elevation of genes involved in immune cell differentiation and function, along with the reduction of genes that facilitate immune cell exhaustion and tolerance[8].
HDAC inhibitors on the market
There are four HDAC inhibitors that have been approved by regulatory agencies and are currently on the market for kinds of cancers.
Vorinostat (Zolinza)
Vorinostat inhibits the activity of class I and II HDAC enzymes, which are involved in the process of histone deacetylation (Figure 2). This inhibition leads to increased histone acetylation and consequent alterations in chromatin structure, ultimately resulting in changes in gene expression. In addition to its effects on histones, Vorinostat can also influence the behavior of non-histone proteins, including transcription factors and chaperone proteins, which can in turn affect gene expression and other cellular processes[10].

Figure 2 Crystal structure of an HDAC homolog complexed with Vorinostat(PDB:1C3S)
Vorinostat has been studied in clinical trials for the treatment of various cancers, including cutaneous T-cell lymphoma (CTCL), multiple myeloma, and solid tumors. In CTCL, Vorinostat has been shown to be effective in improving response rates and progression-free survival. In multiple myeloma, Vorinostat has been tested in combination with other agents and has shown promise in improving response rates and survival. In solid tumors, Vorinostat has shown less effective activity, but studies are ongoing to determine its potential in combination with other agents[11,12]. Common adverse effects of Vorinostat include fatigue, nausea, diarrhea, anorexia, and thrombocytopenia. More serious adverse effects include cardiac toxicity, QT prolongation, and pulmonary embolism. Vorinostat has a narrow therapeutic index, and dose adjustments may be necessary to avoid toxicity. Vorinostat is absorbed rapidly after oral administration, with peak plasma concentrations reached within 2-3 hours[13]. Vorinostat is metabolized primarily by the liver and excreted in the feces. Vorinostat has a short half-life, with a mean elimination half-life of approximately 2 hours. Vorinostat inhibits HDAC activity in a dose-dependent manner, with maximal inhibition achieved at doses of 200-300 mg/m2[14] Beyond its application as an anticancer drug, Vorinostat has been discovered to possess additional capabilities. Among these capabilities is its potential usage in the management of diverse non-cancerous illnesses, including autoimmune and neurodegenerative diseases. Preclinical studies have shown that Vorinostat has anti-inflammatory effects, making it a potential candidate for the treatment of autoimmune diseases such as rheumatoid arthritis and systemic lupus erythematosus. It has been shown to inhibit the production of pro-inflammatory cytokines and chemokines in vitro, and reduce inflammation in animal models of autoimmune diseases[15,16].
Vorinostat has also been studied for its potential use in the treatment of neurodegenerative diseases, such as Huntington’s disease and Alzheimer’s disease. Vorinostat has also been found that have ability to improve cognitive function and reduce motor deficits in animal models of these diseases, possibly by modifying gene expression and improving cellular function[17,18]. Overall, while the primary use of Vorinostat is as an anticancer agent, its potential as a treatment for other diseases is being actively investigated.
Romidepsin (Istodax)
Romidepsin, marketed under the trade name Istodax(Figure 3), is currently approved by the FDA as a histone deacetylase inhibitor (HDACi) for the treatment of patients with cutaneous T-cell lymphoma (CTCL) and peripheral T-cell lymphoma (PTCL) who have received at least one treatment.
Figure 3 The structure of Romidepsin
Romidepsin is a cyclic peptide that was derived from the bacterium Chromobacterium violaceum, and it exerts its anti-cancer effects by inhibiting the activity of class I HDACs, particularly HDAC1 and HDAC2, leading to the accumulation of acetylated histones and the modulation of gene expression[19].
Romidepsin is a class I selective HDAC inhibitor that binds to the catalytic pocket of HDAC enzymes, leading to the accumulation of acetylated histones and the modulation of gene expression. It has been shown to induce growth arrest, differentiation, and apoptosis in cancer cells through the activation of various signaling pathways, including the p21/CDKN1A and p53 pathways, and the upregulation of pro-apoptotic genes such as BAX and FAS. Additionally, Romidepsin has been shown to have anti-angiogenic effects by downregulating the expression of vascular endothelial growth factor (VEGF) and modulating the immune system by increasing the expression of major histocompatibility complex (MHC) class I and II antigens on cancer cells.
Several clinical trials have been conducted to evaluate the efficacy and safety of Romidepsin in the treatment of various cancers. In a phase II study of Romidepsin in patients with relapsed or refractory CTCL, the overall response rate (ORR) was 34%, with a median duration of response of 15 months. In a phase II trial of Romidepsin[20] in patients with PTCL, the ORR was 25%, with a median duration of response of 13.1 months. Romidepsin has been examined in combination with other agents, such as lenalidomide and bortezomib, for treating multiple myeloma and with gemcitabine and cisplatin for treating non-small cell lung cancer. The findings from these studies exhibited favorable outcomes and Romidepsin was generally well-tolerated. Like other HDAC inhibitors, Romidepsin has a range of adverse effects. The most common side effects include nausea, fatigue, thrombocytopenia, anemia, and lymphopenia. In addition, Romidepsin has been reported to cause QT prolongation, which can lead to serious cardiac arrhythmias. Therefore, close monitoring of cardiac function is required during treatment with Romidepsin. The pharmacokinetics of Romidepsin has been studied extensively in both preclinical and clinical settings. Romidepsin is rapidly cleared from the plasma, with a half-life of approximately 3 hours. It is metabolized primarily by CYP3A4 and eliminated mainly through the biliary system. The pharmacodynamic effects of Romidepsin have also been investigated in clinical trials, with evidence of HDAC inhibition observed in peripheral blood mononuclear cells and tumor tissue[21,22].
Romidepsin has the potential to be used not only for cancer treatment but also for non-cancer applications, such as autoimmune diseases and viral infections. Preclinical studies have shown that Romidepsin can impede the replication of various viruses, including Kaposi’s sarcoma-associated herpes virus, Epstein-Barr virus, and HIV. Additionally, Romidepsin has displayed anti-inflammatory properties in animal models of rheumatoid arthritis and multiple sclerosis. Clinical trials are currently underway to evaluate the effectiveness of Romidepsin for these conditions. While Romidepsin is effective in treating various hematological malignancies, it can lead to adverse effects, such as QT prolongation. Romidepsin’s unique structure and mechanism of action may provide advantages over other HDAC inhibitors, and its potential role in cancer immunotherapy and non-cancer indications is still being investigated.
Belinostat (Beleodaq)
Belinostat (Beleodaq) is a histone deacetylase inhibitor that is used for the treatment of peripheral T-cell lymphoma (PTCL), a rare and aggressive form of non-Hodgkin’s lymphoma. It was approved by the US Food and Drug Administration (FDA) in 2014 for this indication.
Figure 4 The structure of Belinostat
Belinostat[23] is a pan-HDAC inhibitor that targets Class I, II, and IV HDAC enzymes. It binds to the active site of HDAC enzymes and blocks their ability to remove acetyl groups from histone proteins. This leads to an accumulation of acetylated histones, which causes chromatin relaxation, and allows for easier access of transcription factors to DNA, leading to changes in gene expression. In addition to histones, HDACs also target non-histone proteins, such as transcription factors and signaling molecules, and Belinostat may affect these proteins as well[24].
Belinostat has been studied in clinical trials[25] for the treatment of various cancers, including PTCL, ovarian cancer, melanoma, and breast cancer, among others. In a phase II study of relapsed or refractory PTCL, Belinostat showed an overall response rate of 25.8%, with a median duration of response of 8.5 months. In a phase II study of ovarian cancer, Belinostat showed modest activity as a single agent, with an overall response rate of 7.1%. However, it showed better activity when used in combination with carboplatin and paclitaxel, with an overall response rate of 27.8%. In a phase II study of melanoma, Belinostat showed some activity as a single agent, with a disease control rate of 27.8%[26]. Belinostat, administered intravenously, exhibits a short half-life of approximately 30 minutes and is rapidly eliminated from the body. Belinostat is metabolized by the liver and excreted via urine, its pharmacodynamics demonstrate dose-dependent inhibition of HDAC activity, with maximum inhibition observed at doses of 1,000 mg/m2. A recent study investigated the impact of Belinostat on the expression of genes implicated in the immune response in patients with autoimmune diseases[27]. Findings indicate that Belinostat stimulated the expression of genes involved in immune regulation while inhibiting genes associated with inflammation, thereby highlighting its potential as a therapy for autoimmune disorders.
Panobinostat (Farydak)
Panobinostat is a histone deacetylase (HDAC) inhibitor used in the treatment of multiple myeloma, a cancer of plasma cells in the bone marrow[28]. It is sold under the brand name Farydak and was approved by the FDA in 2015.
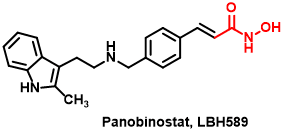
Figure 5 The structure of Panobinostat
Panobinostat is a pan-HDAC inhibitor, meaning it inhibits multiple classes of HDAC enzymes, including Class I, II, and IV HDACs. It functions by blocking the removal of acetyl groups from histone proteins, which results in the relaxation of chromatin structure and the activation of genes that may be involved in cell growth and differentiation[29].
Clinical trials have demonstrated the efficacy of Panobinostat in combination with other anti-cancer drugs in the treatment of relapsed and refractory multiple myeloma. In a phase III clinical trial, the addition of Panobinostat to a standard chemotherapy regimen resulted in a significant improvement in progression-free survival in patients with relapsed multiple myeloma[30]. One study investigated the use of Panobinostat in combination with the immune checkpoint inhibitor nivolumab in patients with advanced solid tumors. The results showed that the combination therapy was well-tolerated and had clinical activity in some patients, noting a potential role for Panobinostat in combination with immunotherapy in the treatment of cancer[31]. Besides its potential for cancer therapy, Panobinostat has also been explored for its possible application in treating other conditions. For example, preclinical research has illustrated its neuroprotective properties in models of Parkinson’s and Huntington’s disease.
Overall, Panobinostat represents an important addition to the armamentarium of drugs used in the treatment of multiple myeloma, and shows promise in the treatment of other types of cancer as well. Ongoing research will continue to elucidate its potential benefits and limitations, and may lead to further refinements in its clinical use. Preclinical studies have suggested that it may also have activity against solid tumors. However, like other HDAC inhibitors, Panobinostat can cause side effects such as fatigue, nausea, diarrhea, and thrombocytopenia[25]. It is important for patients receiving Panobinostat to be closely monitored for these and other potential adverse effects.
Chidamide (Epidaza)
Chidamide[32] has been granted approval in China for its effectiveness in treating relapsed or refractory peripheral T-cell lymphoma (PTCL) by inhibiting histone deacetylase (Figure 6). Moreover, its potential for managing other types of cancer, including lung and breast cancer, is being explored.
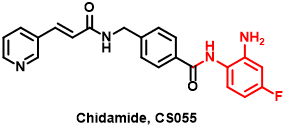
Figure 6 The structure of Chidamide
Chidamide is a novel, orally active inhibitor of histone deacetylase (HDAC) enzymes. By inhibiting HDAC activity, Chidamide promotes acetylation of histones and other non-histone proteins, leading to changes in chromatin structure and gene expression that may affect cell growth and differentiation[33]. Chidamide specifically targets Class I HDACs, which are overexpressed in many types of cancer and are associated with poor prognosis. By inhibiting Class I HDACs, Chidamide induces cell cycle arrest and apoptosis in cancer cells, while sparing normal cells. Chidamide has also been shown to have anti-angiogenic and immunomodulatory effects, which may contribute to its anti-cancer activity. Chidamide has been evaluated in numerous clinical trials for the treatment of various solid tumors.
In a phase II study in patients with advanced non-small cell lung cancer (NSCLC)[34], Chidamide monotherapy showed modest efficacy, with an objective response rate (ORR) of 7.7% and a disease control rate (DCR) of 53.8%. In a phase II trial in patients with relapsed or refractory peripheral T-cell lymphoma (PTCL)[35], Chidamide monotherapy resulted in an ORR of 28.6% and a DCR of 71.4%. Chidamide has also been investigated in combination with chemotherapy or targeted therapies in various solid tumors. In a phase Ib trial in patients with advanced gastric cancer, Chidamide plus chemotherapy (paclitaxel and cisplatin) resulted in an ORR of 53.3% and a DCR of 93.3%. In a phase Ib study in patients with hepatocellular carcinoma (HCC), Chidamide plus sorafenib (a multi-kinase inhibitor) showed promising activity, with an ORR of 35.3% and a DCR of 82.4%[36]. While some studies suggest that Chidamide may have potential as a treatment for autoimmune diseases, further clinical trials are needed to confirm its efficacy and safety in these indications.
To sum up, Chidamide exhibits promising potential as an HDAC inhibitor in treating solid tumors, especially hematological malignancies. Clinical studies have demonstrated its effectiveness when combined with other anti-cancer drugs in treating different types of cancers such as lymphoma, breast cancer, and sarcoma. However, more research is necessary to comprehensively comprehend Chidamide’s mechanisms of action and its possibilities in treating both cancer and non-cancer-related illnesses.
Recent research of HDAC inhibitors
In recent years, considerable progress has been made in the design and creation of HDAC inhibitors[37]. One notable area of research has centered on the creation of potent and selective inhibitors capable of targeting individual HDAC isoforms. This strategy holds promise for enhancing the effectiveness and safety of these medications, given that distinct HDAC isoforms play unique roles in regulating gene expression.
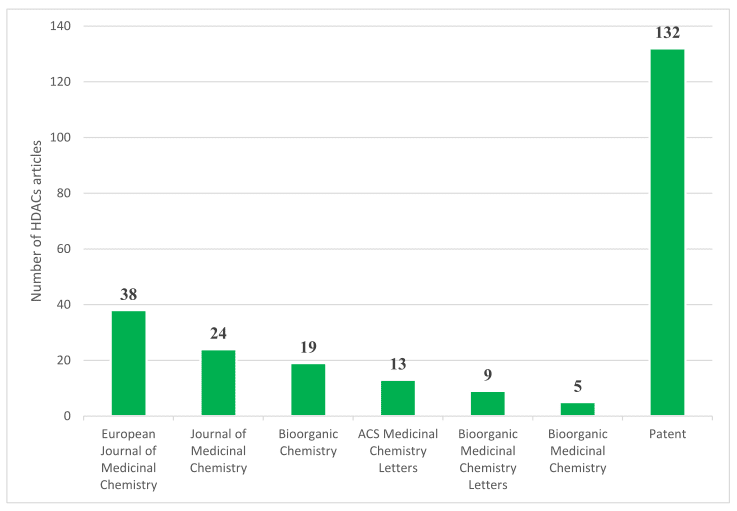
Figure 7 The distribution of HDACs publication in major medicinal chemistry journals and patents since 2020[37].
Structure-activity relationships (SAR) studies have played a critical role in the design and optimization of HDAC inhibitors. SAR studies involve the systematic modification of a lead compound to determine how changes in its chemical structure affect its biological activity[38]. These studies have been used to identify key structural features that are essential for HDAC inhibition, as well as to optimize the pharmacokinetic and pharmacodynamic properties of these compounds. One important development in HDAC inhibitor design has been the incorporation of zinc-binding groups (ZBGs) that can chelate with the active site zinc ion in HDACs. The choice of ZBG can significantly impact the potency, selectivity, and pharmacokinetic properties of HDAC inhibitors. For example, hydroxamic acids have been widely used as ZBGs due to their high affinity for zinc ions and excellent HDAC inhibitory activity. However, hydroxamic acids can also exhibit poor selectivity and cause off-target effects. Alternative ZBGs, such as carboxylic acids and mercaptoacetamides, have been explored as potential alternatives to hydroxamic acids. Another important factor that can impact the activity and selectivity of HDAC inhibitors is the length and flexibility of the linker that connects the ZBG to the effector domain of the molecule. The linker can affect the orientation of the ZBG in the active site of HDACs and thus impact the potency and selectivity of the inhibitor. For example, short, rigid linkers have been shown to increase the potency of HDAC inhibitors by promoting the formation of a stable inhibitor-HDAC complex. However, longer and more flexible linkers can improve the selectivity of HDAC inhibitors by allowing them to adopt different conformations and interact with different amino acid residues in the active site.
The SAR studies and the development of HDAC inhibitors has also been aided by advances in computational chemistry and molecular modeling[39]. Molecular docking, molecular dynamics simulations, and free energy calculations have been utilized to forecast the binding modes of HDAC inhibitors and to develope new compounds with enhanced potency and selectivity. These computational methods have also been employed to explore the structural and dynamic characteristics of HDACs and to decipher the molecular mechanisms of HDAC inhibition. A recent trend in HDAC inhibitor research is the application of covalent inhibitors, which are capable of forming a covalent bond with the active site residue of HDACs. Covalent inhibitors can provide enhanced potency and selectivity by forming a stable, irreversible complex with the target protein. However, the use of covalent inhibitors can also raise concerns about off-target effects and potential toxicity.
Finally, the development of HDAC inhibitors with dual or multiple targets gets more and more interest in recent years. These compounds can target both HDACs and other proteins involved in disease pathogenesis, such as kinases, epigenetic readers, and transcription factors. Dual-targeting HDAC inhibitors have the potential to provide enhanced efficacy by targeting multiple pathways involved in disease progression. However, the design of dual-targeting HDAC inhibitors is difficult, as it requires the design of two or more distinct pharmacophores within a single molecule.
In terms of selectivity, recent research has focused on developing inhibitors that can target specific HDAC isoforms. HDAC isoforms play distinct roles in regulating gene expression, and targeting specific isoforms can potentially improve the efficacy and reduce the side effects of HDAC inhibitors. One approach to achieve isoform-selective inhibition is the use of substrate-based inhibitors, which mimic the natural substrates of specific HDAC isoforms and target their unique active site features. For example, a substrate-based inhibitor of HDAC6 was recently reported that showed excellent selectivity for HDAC6 over other HDAC isoforms. Another approach to achieving isoform-selective inhibition is the use of allosteric modulators that can bind to non-active site regions of HDACs and regulate their activity. Allosteric modulators can provide selective inhibition by targeting the unique conformational changes and regulatory mechanisms of specific HDAC isoforms. For example, a recent study reported the identification of an allosteric modulator of HDAC3 that showed excellent selectivity and efficacy in preclinical models of cancer.
In addition to targeting specific HDAC isoforms, there has been interest in developing HDAC inhibitors that can penetrate the blood-brain barrier (BBB) and target HDACs in the central nervous system (CNS). The BBB is a complex network of blood vessels that separates the CNS from the peripheral circulation and presents a significant challenge for the development of CNS-targeted drugs. Several strategies have been explored to overcome this challenge, including the use of prodrugs that can be converted to active compounds within the CNS, the modification of HDAC inhibitors with BBB-penetrating moieties, and the development of nanoparticles that can transport HDAC inhibitors across the BBB.
In summary, recent advances in the design and development of HDAC inhibitors have focused on improving their potency, selectivity, and pharmacokinetic properties. These advances have been driven by a combination of SAR studies, computational modeling, and innovative drug discovery approaches. The development of highly potent and selective HDAC inhibitors that can target specific isoforms or penetrate the BBB has the potential to significantly enhance the efficacy and safety of these drugs in the treatment of cancer, neurological disorders, and other diseases[40].
Summary
HDACs play a critical role in the regulation of gene expression and cellular processes in the human body. The dysregulation of HDAC activity has been implicated in the development and progression of various diseases, including cancer, neurodegeneration, and metabolic disorders. HDAC inhibitors are a promising therapeutic strategy for the treatment of these diseases, as they can modulate gene expression and cellular processes by inhibiting HDAC activity. HDAC inhibitors work by disrupting the histone code, protein-protein interactions, and immune response, and inducing DNA damage while activating the DNA damage response pathway. Nonetheless, more research is required to gain a complete understanding of the mechanisms of HDAC inhibitor action and to produce more effective and specific HDAC inhibitors for clinical use.
Reference
Choudhary, C., Kumar, C., Gnad, F., Nielsen, M. L., Rehman, M., Walther, T. C., … & Mann, M. (2009). Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science, 325(5942), 834-840.
Glozak, M. A., Sengupta, N., Zhang, X., & Seto, E. (2005). Acetylation and deacetylation of non-histone proteins. gene, 363, 15-23.
Minucci, S., & Pelicci, P. G. (2006). Histone deacetylase inhibitors and the promise of epigenetic (and more) treatments for cancer. Nature Reviews Cancer, 6(1), 38-51.
Marks, P. A., Richon, V. M., Breslow, R., & Rifkind, R. A. (2001). Histone deacetylase inhibitors as new cancer drugs. Current opinion in oncology, 13(6), 477-483.
Bolden, J. E., Peart, M. J., & Johnstone, R. W. (2006). Anticancer activities of histone deacetylase inhibitors. Nature reviews Drug discovery, 5(9), 769-784.
Jenuwein, T., & Allis, C. D. (2001). Translating the histone code. Science, 293(5532), 1074-1080.
Jackson, S. P., & Bartek, J. (2009). The DNA-damage response in human biology and disease. Nature, 461(7267), 1071-1078.
West, A. C., & Johnstone, R. W. (2014). New and emerging HDAC inhibitors for cancer treatment. The Journal of clinical investigation, 124(1), 30-39.
Richon, V. M., Sandhoff, T. W., Rifkind, R. A., & Marks, P. A. (2000). Histone deacetylase inhibitor selectively induces p21WAF1 expression and gene-associated histone acetylation. Proceedings of the National Academy of Sciences, 97(18), 10014-10019.
Kelly, W. K., Richon, V. M., O’Connor, O., Curley, T., MacGregor-Curtelli, B., Tong, W., … & Scher, H. (2003). Phase I clinical trial of histone deacetylase inhibitor: suberoylanilide hydroxamic acid administered intravenously. Clinical Cancer Research, 9(10), 3578-3588.
Marks, P. A., & Breslow, R. (2007). Dimethyl sulfoxide to vorinostat: development of this histone deacetylase inhibitor as an anticancer drug. Nature biotechnology, 25(1), 84-90.
Mann, B. S., Johnson, J. R., Cohen, M. H., Justice, R., & Pazdur, R. (2007). FDA approval summary: vorinostat for treatment of advanced primary cutaneous T-cell lymphoma. The oncologist, 12(10), 1247-1252.
Marks, P. A., Richon, V. M., & Rifkind, R. A. (2000). Histone deacetylase inhibitors: inducers of differentiation or apoptosis of transformed cells. Journal of the National Cancer Institute, 92(15), 1210-1216.
Butler, L. M., Agus, D. B., Scher, H. I., Higgins, B., Rose, A., Cordon-Cardo, C., … & Richon, V. M. (2000). Suberoylanilide hydroxamic acid, an inhibitor of histone deacetylase, suppresses the growth of prostate cancer cells in vitro and in vivo. Cancer research, 60(18), 5165-5170.
Grabiec, A. M., Korchynskyi, O., Tak, P. P., & Reedquist, K. A. (2012). Histone deacetylase inhibitors suppress rheumatoid arthritis fibroblast-like synoviocyte and macrophage IL-6 production by accelerating mRNA decay. Annals of the rheumatic diseases, 71(3), 424-431.
Ferrante, R. J., Kubilus, J. K., Lee, J., Ryu, H., Beesen, A., Zucker, B., … & Hersch, S. M. (2003). Histone deacetylase inhibition by sodium butyrate chemotherapy ameliorates the neurodegenerative phenotype in Huntington’s disease mice. Journal of Neuroscience, 23(28), 9418-9427.
Ricobaraza, A., Cuadrado‐Tejedor, M., Marco, S., Pérez‐Otaño, I., & García‐Osta, A. (2012). Phenylbutyrate rescues dendritic spine loss associated with memory deficits in a mouse model of Alzheimer disease. Hippocampus, 22(5), 1040-1050.
Prince, H. M., Dickinson, M., & Khot, A. (2013). Romidepsin for cutaneous T-cell lymphoma. Future Oncology, 9(12), 1819-1827.
Piekarz, R. L., Frye, R., Prince, H. M., Kirschbaum, M. H., Zain, J., Allen, S. L., … & Bates, S. E. (2011). Phase 2 trial of romidepsin in patients with peripheral T-cell lymphoma. Blood, The Journal of the American Society of Hematology, 117(22), 5827-5834.
Connolly, R. M., Laille, E., Vaishampayan, U., Chung, V., Kelly, K., Dowlati, A., … & Rudek, M. A. (2020). Phase I and pharmacokinetic study of romidepsin in patients with cancer and hepatic dysfunction: a National Cancer Institute Organ Dysfunction Working Group Study. Clinical Cancer Research, 26(20), 5329-5337.
Foss, F., Horwitz, S., Pro, B., Prince, H. M., Sokol, L., Balser, B., … & Coiffier, B. (2016). Romidepsin for the treatment of relapsed/refractory peripheral T cell lymphoma: prolonged stable disease provides clinical benefits for patients in the pivotal trial. Journal of hematology & oncology, 9(1), 1-8.
Tan, J., Cang, S., Ma, Y., Petrillo, R. L., & Liu, D. (2010). Novel histone deacetylase inhibitors in clinical trials as anti-cancer agents. Journal of hematology & oncology, 3(1), 1-13.
Plumb, J. A., Finn, P. W., Williams, R. J., Bandara, M. J., Romero, M. R., Watkins, C. J., … & Brown, R. (2003). Pharmacodynamic response and inhibition of growth of human tumor xenografts by the novel histone deacetylase inhibitor PXD101. Molecular cancer therapeutics, 2(8), 721-728.
Prince, H. M., Bishton, M. J., & Harrison, S. J. (2009). Clinical studies of histone deacetylase inhibitors. Clinical Cancer Research, 15(12), 3958-3969.
O’Connor, O. A., Horwitz, S., Masszi, T., Van Hoof, A., Brown, P., Doorduijn, J., … & Shustov, A. (2015). Belinostat in patients with relapsed or refractory peripheral T-cell lymphoma: results of the pivotal phase II BELIEF (CLN-19) study. Journal of Clinical Oncology, 33(23), 2492.
Zhang, X. H., Qin-Ma, Wu, H. P., Khamis, M. Y., Li, Y. H., Ma, L. Y., & Liu, H. M. (2021). A review of progress in histone deacetylase 6 inhibitors research: structural specificity and functional diversity. Journal of medicinal chemistry, 64(3), 1362-1391.
Dokmanovic, M., Clarke, C., & Marks, P. A. (2007). Histone deacetylase inhibitors: overview and perspectives. Molecular cancer research, 5(10), 981-989.
Atadja, P. (2009). Development of the pan-DAC inhibitor panobinostat (LBH589): successes and challenges. Cancer letters, 280(2), 233-241.
San-Miguel, J. F., Hungria, V. T., Yoon, S. S., Beksac, M., Dimopoulos, M. A., Elghandour, A., … & Richardson, P. G. (2014). Panobinostat plus bortezomib and dexamethasone versus placebo plus bortezomib and dexamethasone in patients with relapsed or relapsed and refractory multiple myeloma: a multicentre, randomised, double-blind phase 3 trial. The lancet oncology, 15(11), 1195-1206.
Suraweera, A., O’Byrne, K. J., & Richard, D. J. (2018). Combination therapy with histone deacetylase inhibitors (HDACi) for the treatment of cancer: achieving the full therapeutic potential of HDACi. Frontiers in oncology, 8, 92.
Lu, X., Ning, Z., Li, Z., Cao, H., & Wang, X. (2016). Development of chidamide for peripheral T-cell lymphoma, the first orphan drug approved in China. Intractable & rare diseases research, 5(3), 185-191.
Ning, Z. Q., Li, Z. B., Newman, M. J., Shan, S., Wang, X. H., Pan, D. S., … & Lu, X. P. (2012). Chidamide (CS055/HBI-8000): a new histone deacetylase inhibitor of the benzamide class with antitumor activity and the ability to enhance immune cell-mediated tumor cell cytotoxicity. Cancer chemotherapy and pharmacology, 69, 901-909.
Zhou, Y., Pan, D. S., Shan, S., Zhu, J. Z., Zhang, K., Yue, X. P., … & Ning, Z. Q. (2014). Non-toxic dose chidamide synergistically enhances platinum-induced DNA damage responses and apoptosis in non-small-cell lung cancer cells. Biomedicine & Pharmacotherapy, 68(4), 483-491.
Shi, Y., Dong, M., Hong, X., Zhang, W., Feng, J., Zhu, J., … & Lu, X. (2015). Results from a multicenter, open-label, pivotal phase II study of chidamide in relapsed or refractory peripheral T-cell lymphoma. Annals of oncology, 26(8), 1766-1771.
Dong, M., Ning, Z. Q., Xing, P. Y., Xu, J. L., Cao, H. X., Dou, G. F., … & Feng, F. Y. (2012). Phase I study of chidamide (CS055/HBI-8000), a new histone deacetylase inhibitor, in patients with advanced solid tumors and lymphomas. Cancer chemotherapy and pharmacology, 69, 1413-1422.
He, X., Hui, Z., Xu, L., Bai, R., Gao, Y., Wang, Z., … & Ye, X. Y. (2022). Medicinal chemistry updates of novel HDACs inhibitors (2020 to present). European Journal of Medicinal Chemistry, 227, 113946.
Patel, V. K., Shirbhate, E., Tiwari, P., Kore, R., Veerasamy, R., Mishra, A., & Rajak, H. (2023). Multi-targeted HDAC Inhibitors as Anticancer Agents: Current Sta-tus and Future Prospective. Current Medicinal Chemistry.
Avelar, L. A. A., Schrenk, C., Sönnichsen, M., Hamacher, A., Hansen, F. K., Schliehe-Diecks, J., … & Kurz, T. (2021). Synergistic induction of apoptosis in resistant head and neck carcinoma and leukemia by alkoxyamide-based histone deacetylase inhibitors. European Journal of Medicinal Chemistry, 211, 113095.
Stott, A. J., Maillard, M. C., Beaumont, V., Allcock, D., Aziz, O., Borchers, A. H., … & Dominguez, C. (2021). Evaluation of 5-(Trifluoromethyl)-1, 2, 4-oxadiazole-based class IIa HDAC inhibitors for Huntington’s disease. ACS Medicinal Chemistry Letters, 12(3), 380-388.