Latest Progress in AI Drug Development
Abstract
Drug development is an inherently complex and protracted endeavor, traditionally guided by expert intuition and iterative trial-and-error experimentation. However, the advent of artificial intelligence (AI)—particularly large language models and generative AI—heralds a transformative shift in this domain. The integration of AI-driven methodologies into various stages of the drug development pipeline is already yielding subtle yet significant gains in both efficiency and effectiveness. This article offers a comprehensive overview of recent advancements in AI applications throughout the drug development continuum, encompassing target identification, compound discovery, preclinical and clinical evaluation, as well as post-marketing surveillance. Furthermore, we critically analyze the prevailing challenges and outline prospective directions for future research in AI-augmented drug development.
Introduction
Drug development is a highly intricate and multifaceted endeavor aimed at creating new therapeutics to combat a wide range of diseases. The process encompasses several critical stages, including target identification, compound discovery, preclinical testing, clinical trials, regulatory approval, and post-marketing surveillance. Despite its importance, modern drug development faces significant hurdles—most notably exorbitant costs, prolonged timelines, and a strikingly low success rate. On average, bringing a single drug to market demands an investment of approximately $2.6 billion and spans 12 to 15 years. Alarmingly, the probability of a drug successfully advancing through clinical trials remains below 10%.
Several factors underlie these challenges. Many diseases are inherently complex and multifactorial, complicating the search for effective therapeutic targets. The drug development pipeline itself is layered and susceptible to failure at any stage, which can compromise the entire effort. Moreover, the chemical space that must be navigated to identify viable drug candidates is vast—estimated between 10⁶⁰ and 10¹⁰⁰ possible molecules—rendering drug discovery akin to searching for a needle in a haystack. Additionally, stringent regulatory standards require comprehensive evidence of safety, efficacy, and quality, further extending development timelines and escalating costs.
In response to these obstacles, researchers are increasingly turning to innovative technologies to enhance and streamline the drug development process. Among these, artificial intelligence (AI) has emerged as a particularly promising tool with the potential to revolutionize the field.
Recent advances in artificial intelligence (AI), including image recognition, natural language processing (NLP), and computer vision, have shown particular promise in addressing key challenges in drug development. In particular, large language models (LLMs) such as ChatGPT, DeepSeek, Grok, and Gemini, as well as generative AIs such as Sora, have demonstrated the ability to surpass human intelligence in certain situations. AI’s ability to process vast amounts of data has the potential to greatly accelerate and improve the drug development process. As a result, pharmaceutical companies, biotechnology companies, and research institutions are increasingly adopting AI-driven approaches to overcome the obstacles inherent in traditional approaches. AI has proven to be valuable in analyzing complex biological systems, identifying disease biomarkers and potential drug targets, modeling drug-target interactions, predicting the safety and efficacy of drug candidates, and managing clinical trials (Figure 1). However, it is important to recognize that AI-driven drug development still faces some unique challenges. If these obstacles are not effectively addressed, the potential of AI may not be fully realized.
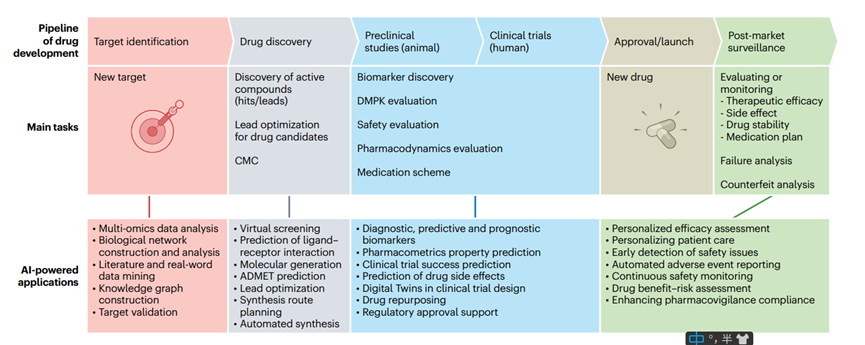
Figure 1. AI integration in drug development pipeline
This review explores the most advanced applications of artificial intelligence in small molecule drug development since 2019. In this review, we first describe AI-driven drug discovery, from target identification to synthesis planning, and the application of AI in the clinical phase of drug development – including biomarker discovery, drug repurposing, prediction of pharmacokinetic properties and toxicity, and the conduct of clinical trials. Finally, we discuss the challenges facing AI-driven drug development and outline future directions in the field. We hope to shed light on a new era of innovation, efficiency, and precision in drug development, which will hopefully accelerate the delivery of new and better medicines to patients.
AI-driven drug discovery
In recent years, AI has become a transformative force in drug discovery, revolutionizing traditional methods and improving efficiency at multiple stages of the process. This section will explore the profound impact of AI on various aspects of drug discovery, including target identification, virtual screening, de novo design, ADMET (absorption, distribution, metabolism, excretion, and toxicity) prediction, as well as synthesis planning and automated synthesis and drug discovery. By leveraging advanced algorithms and technologies, researchers are now able to accelerate the discovery of new therapeutics, improve the accuracy of predictions, and reduce the overall time and cost associated with drug development.
Target Identification
The identification of small molecule targets (such as proteins or nucleic acids) is a key process in drug discovery. Traditional methods such as affinity pull-down assays and genome-wide knockout screening are widely used, but are often time-consuming and labor-intensive with high failure rates. Advances in artificial intelligence technology are revolutionizing this field by analyzing large datasets in complex biological networks. Artificial intelligence facilitates the identification of disease-associated molecular patterns and causal relationships by constructing multi-omics data networks, thereby driving the discovery of candidate drug targets. For example, recent studies have used natural language processing techniques (such as word2vec embedding) to map gene functions into high-dimensional space, improving the sensitivity of target identification despite sparse gene function overlap. However, effectively integrating multi-omics data and ensuring the interpretability of artificial intelligence models remain challenging tasks. Graph deep learning techniques address these issues by combining graph structures with deep learning to focus on graph nodes associated with key features (e.g., atom type, charge) to effectively identify candidate targets. A recent study successfully developed an interpretable framework using multi-omics network graphs with graph attention mechanisms to effectively predict cancer genes. In addition, integrating multi-omics data with scientific and medical literature into knowledge graphs enables AI to identify relationships between genes and disease pathways. When biomedical large language models are deeply integrated with biological network or knowledge graph capabilities, they provide an efficient and accurate method for connecting diseases, genes, and biological processes.
Real-world data, such as medical records, self-reports, electronic health records (EHRs), and insurance claims, provide important contextual information for understanding complex diseases and facilitating target discovery. However, real-world data often contain unstructured text, lack standardization, and may contain biases, which limits their application in this context. While high-quality, curated datasets are essential for training models, real-world data are inherently noisy and complicated by the confluence of multiple diseases. Despite these issues, recent studies have demonstrated that noisy real-world data can still train effective models, thus improving the potential for gene discovery and candidate drug targets in scenarios with noisy medical records and non-expert disease labels. Enhancing the generalization ability of models across different populations remains a major challenge, especially for diseases with poor labels or prevalence. As real-world and multi-omics data become increasingly abundant, leveraging advanced data mining algorithms and expert knowledge will further enhance their integration and significantly improve the success rate of target discovery.
Virtual screening
Virtual screening is a key strategy for efficiently identifying potential lead compounds or drug candidates. The rapid expansion of compound libraries requires accelerated virtual screening of very large libraries, which has driven the development of ligand docking artificial intelligence technology. AI-based receptor-ligand docking models can predict spatial transformations of ligands, directly generate complex atomic coordinates using algorithms such as equivariant neural networks, and learn probability density distributions of receptor-ligand distances to generate binding poses. Notably, the latest receptor-ligand co-folding networks based on AlphaFold2 and RosettaFold show promise in predicting complex structures directly from sequence information. However, due to insufficient learning of physical constraints, they may produce unrealistic ligand conformations, so post-processing (e.g., energy minimization) or geometric constraints are required to optimize the effectiveness of docking poses. However, in pocket-oriented docking tasks, deep learning-based binding pose prediction models have not yet surpassed physics-based methods, and they usually do not fully consider the flexibility of receptor pockets. In addition, predicting accurate receptor-ligand interactions remains a challenge. While early machine learning successes in affinity prediction have stimulated interest in deep learning models, and these models can outperform traditional scoring functions by processing three-dimensional structural and non-structural data, their performance depends heavily on the accuracy of the ligand pose and is primarily applicable to known receptor structures.
Direct application of docking-based virtual screening is impractical when the target structure is missing or incomplete. As an alternative, artificial intelligence techniques can be used for sequence-based prediction methods. However, these methods often have difficulty capturing the complexity of three-dimensional protein-ligand interactions, complicating accurate predictions of how changes in binding poses affect interaction strength.
While targeted drug development is effective for well-defined targets, many diseases lack such targets. Therefore, phenotype-based virtual screening is essential for diseases with unclear targets (e.g., rare diseases) and broad phenotypic diseases (e.g., aging). A recent study used nuclear morphology and machine learning to identify compounds that induce senescence in cancer cells; similar strategies are also promising in antibiotic discovery. However, such models often rely on case-specific phenotypic data and have difficulties in generalization. In addition, artificial intelligence activity predictions that rely solely on ligand chemical structure face challenges such as sparse and unbalanced data and activity cliffs. Recent studies have shown that integrating relevant biological information such as cell morphology and transcriptional profiles can improve model performance, providing a new direction for more accurate activity predictions.
Current virtual screening models often focus on specific tasks such as scoring, pose optimization, or screening, which highlights the need to develop general models that can handle multiple tasks. Incorporating inductive bias (the inherent tendency of a model to prioritize certain types of solutions over others) or data augmentation (techniques used to artificially expand the diversity of training datasets without collecting new data) may improve the generalization ability of models. In addition, commercial compound collections are growing exponentially to billions, making comprehensive screening computationally infeasible. At the same time, available molecular libraries cover only a small portion of the druggable chemical space, which is still expanding – this brings opportunities and challenges to navigating and screening bioactive molecules.
To address these challenges, techniques such as active learning and Bayesian optimization are effective methods to solve the chemical space search problem and become key to improving the efficiency of virtual screening. Combining quantum mechanics with artificial intelligence provides new tools for chemical space exploration, while molecular dynamics simulations deepen the understanding of protein-ligand interactions and address binding affinity and selectivity issues, thereby improving model accuracy. At the same time, deep generative models significantly reduce the search space and improve screening efficiency by generating customized virtual libraries for specific targets or compound types. For example, the conditional recurrent neural network we developed generated a custom library that identified a potent and selective RIPK1 inhibitor in cell and animal models.
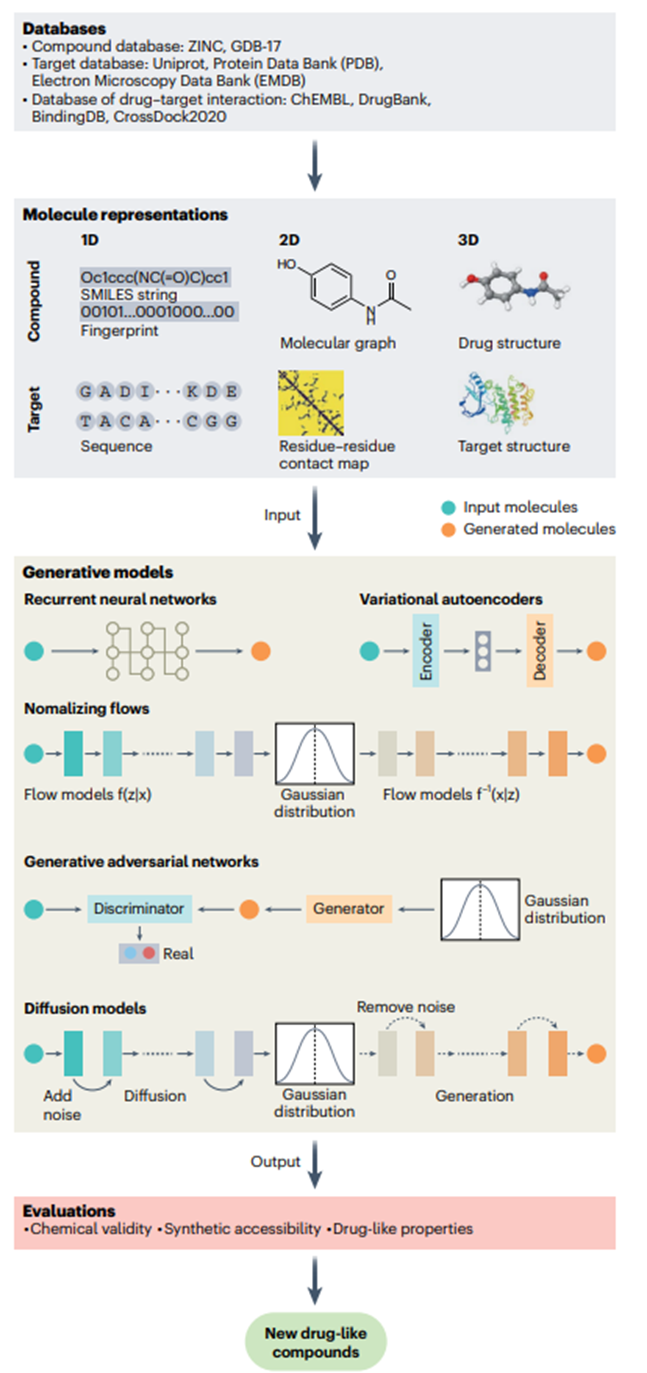
Figure 2. AI molecule generation process
De novo design
De novo drug design involves the autonomous creation of new chemical structures that optimally meet desired molecular features. Traditional structure-based, ligand-based, and pharmacophore-based design methods are manual and rely on expert designers and explicit rules. Artificial intelligence, especially deep learning, enables the automatic identification of new structures that meet specific requirements, bypassing traditional expertise. This technology has been successfully applied to the development of small molecule inhibitors, PROTACs, peptides, and functional proteins validated by wet lab experiments, ushering in a more efficient and innovative era of drug discovery.
In deep learning-driven de novo design (Figure 2), the molecule generation component is the core, usually using chemical language or graph-based models. Chemical language models transform the molecule generation task into sequence generation, such as SMILES strings (“Simplified Molecular Encoding Line Encoding System”, a representation system that represents chemical structures in a linear text format). Although a lot of pre-training is required and invalid SMILES may be generated due to syntax errors, these errors can help the model self-correct by filtering unlikely samples. Models such as the Long Short-Term Memory model (a deep learning model for analyzing sequence data) face information compression bottlenecks that hinder the learning of global sequence properties, suggesting the need for architectures such as Transformers to better capture global properties. Recent work has integrated structured state-space sequences into chemical language models, revealing high chemical space similarities and alignment with key natural product design features, demonstrating the usefulness of this model for de novo design.
In contrast, graph-based models represent molecules as graphs and generate structures using autoregressive or non-autoregressive strategies. Autoregressive methods build molecules atom by atom, which can lead to chemically implausible intermediates and introduce biases. In contrast, non-autoregressive methods generate the entire molecular graph at once, but require additional steps to ensure the validity of the graph, as these models’ limited perception of the molecular topology can lead to flawed structures.
Given the vastness of drug-like chemical space, de novo generation often uses optimization mechanisms such as scoring functions based on metrics including similarity to known active molecules and predicted bioactivity to guide the design toward target features. Incorporating reinforcement learning for iterative optimization is an effective approach, but designing appropriate scoring functions is challenging because directly quantifying objectives such as synthetic feasibility or drug-likeness is difficult and often leads to unintended consequences. In addition, the extensive optimization steps of reinforcement learning highlight challenges in sample efficiency that may be mitigated by active or curriculum learning strategies.
In addition to introducing scoring functions, incorporating constraints—such as disease-related gene expression signatures, pharmacophores, protein sequence or structure, binding affinity, and protein-ligand interactions—can also guide the model to generate desired molecules. For example, the PocketFlow model, conditional on protein pockets, effectively generated experimentally validated active compounds against HAT1 and YTHDC1 targets, demonstrating its drug design capabilities. In addition, the model can optimize lead compounds by restricting the output to specific scaffolds or fragments of desired candidates, although this comes at the expense of limiting chemical diversity.
ADMET
ADMET plays a crucial role in determining drug efficacy and safety. While wet lab evaluation is required for market approval and cannot be completely replaced by simulations, early ADMET predictions can help reduce failures due to poor properties. Artificial intelligence has emerged as a valuable tool for predicting ADMET properties using predefined features such as molecular fingerprints or descriptors. For example, Bayer’s in silico ADMET platform uses machine learning techniques such as random forests and support vector machines and connects descriptors such as fingerprints using cyclic expansion to ensure accuracy and relevance. Various descriptors have been developed over the past few decades for ADMET prediction. However, the feature engineering involved in these feature-based approaches remains complex and limits versatility and flexibility.
Deep learning now drives ADMET prediction, automatically extracting meaningful features from simple input data. Various neural network architectures, including Transformers (designed to efficiently process sequence data), convolutional neural networks (a type of deep learning model commonly used for image and video recognition tasks), and more recently graph neural networks (deep learning models for processing graph-structured data such as molecular structures), are adept at modeling molecular properties from formats such as SMILES strings and molecular graphs. SMILES strings provide a compact molecular representation and can clearly express substructures such as branching, rings, and chirality, but lack topological awareness – while graph neural networks (such as GeoGNN models) incorporate geometric information, providing superior performance in ADMET prediction. In fact, a recent study showed that Transformer models using SMILES inputs have difficulty in full structure recognition. For predictions involving properties such as toxicity, the performance of representations generated by these models may saturate before training progresses, showing limited improvement after training.
Despite advances driven by new deep learning algorithms, the field still faces challenges. High costs and significant time investment lead to a scarcity of labeled data in ADMET prediction, resulting in potential overfitting. Unsupervised and self-supervised learning provide solutions. Although large Transformer-based models have shown promise in other fields, their application in ADMET prediction remains underexplored. A recent study showed that although the SMILES language does not directly encode molecular topology, carefully designed self-supervised training using a contextual Transformer equipped with a linear attention mechanism can effectively learn implicit structure-property relationships, thereby enhancing the confidence in applying large-scale self-supervised models to ADMET prediction.
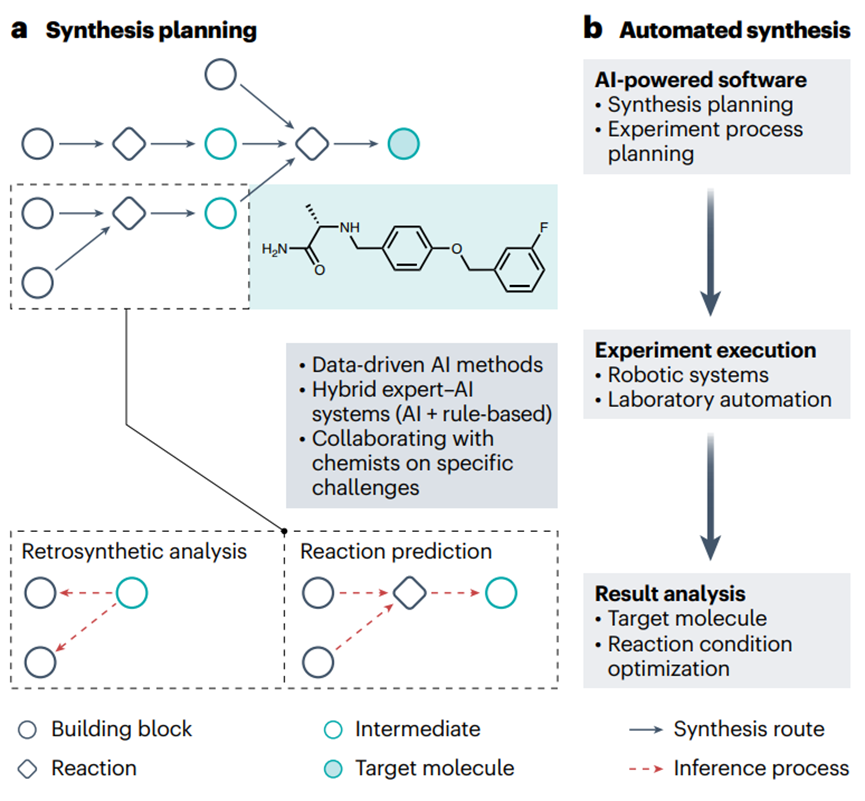
Figure 3. AI-driven drug synthesis prediction
Furthermore, molecular representation is critical to the performance of AI. High-dimensional representations generally provide richer information than low-dimensional representations. However, recent studies have shown that integrating multiple levels of molecular representation can significantly enhance learning, resulting in more comprehensive, more generalizable, and more robust ADMET predictions. This suggests that multimodal ADMET models that use multiple representations simultaneously have potential, although the optimal combination of data types remains unresolved. Interpretability remains a major challenge. Understanding model parameters in ADMET predictions helps reveal the relationship between molecular substructures and properties. Attention mechanisms, which allow models to focus on important parts of the input data, can enhance interpretability by identifying key atoms or groups. Integrating chemical knowledge can further enhance interpretability, but scaling models to achieve comprehensive chemical understanding remains challenging.
Synthesis planning and automated synthesis and drug discovery
Chemical synthesis is one of the bottlenecks in small molecule drug discovery and is a highly technical and extremely tedious task. Computer-assisted synthesis planning (CASP) and automated synthesis of organic compounds help relieve chemists of the burden of repetitive and tedious work, allowing them to engage in more innovative work. With the rapid development of artificial intelligence, the pharmaceutical industry and academia are increasingly interested in making this process intelligent and automated.
CASP has been used as a tool to assist chemists in determining reaction routes through retrosynthetic analysis, a problem-solving technique in which target molecules are recursively converted into increasingly simpler precursors (Figure 3a). Early CASP programs were rule-based (e.g., logic and heuristic methods applied to synthesis analysis, chemical synthesis simulation and evaluation, and retrosynthesis-based synthetic accessibility assessment programs). Since then, a series of machine learning techniques, especially deep learning models, have been developed—making step-by-step improvements in the synthesis planning of artificial small molecules and natural products. Recently, Transformer models have also been applied to retrosynthetic analysis, prediction of regioselectivity (the preference of a chemical reaction to occur at one specific position over another on a molecule with multiple possible reaction sites) and stereoselectivity (the preference of a reaction to produce one stereoisomer over another when multiple stereoisomeric products are present), and reaction fingerprint extraction. However, the adequacy of purely data-driven AI approaches for complex synthetic planning has prompted the development of hybrid expert-AI systems that incorporate chemical rules. However, most current deep learning methods are not interpretable and appear as “black boxes” that provide limited insights. To address this challenge, a new retrosynthetic prediction model, RetroExplainer, was recently introduced with an interpretable deep learning framework that reframes the retrosynthetic task as a molecular assembly process. RetroExplainer shows superior performance compared to state-of-the-art retrosynthetic methods. Notably, its molecular assembly approach enhances interpretability, enabling transparent decision making and quantitative attribution.
Automated synthesis of organic compounds represents a frontier in chemistry-related fields, including medicinal chemistry (Figure 3b). An optimal automated synthesis platform will seamlessly integrate and streamline the various components of the chemical development process, including CASP and automated experimental setup and optimization, as well as robotically performed chemical synthesis, separation, and purification. Recently, deep learning-driven automated flow chemistry and solid-phase synthesis technologies for the synthesis of drug compounds have received considerable attention. In particular, automated synthesis is combined with design, test, and analysis technologies to form the automated central process of drug discovery, known as the design-make-test-analyze (DMTA) cycle. By leveraging deep learning, the efficiency of the DMTA cycle has been significantly improved, accelerating the discovery of lead compounds for drug discovery. For example, a liver X receptor agonist was generated de novo by using an AI-driven DMTA platform that leverages deep learning for molecular design and microfluidics for on-chip chemical synthesis. In addition, large language models (LLMs) are thought to “understand” human natural language, enabling automated platforms to provide tailored solutions to specific challenges based on researchers’ concise input. Despite the promise of automated synthesis and automated DMTA cycles, their development is still in its infancy. Many technical challenges remain, including reducing solid formation to avoid clogging, predicting solubility in non-aqueous solvents and at different temperatures, estimating optimal purification methods, and optimizing requirements for multi-step reactions.
After the planning and synthesis of new drug compounds, AI technologies facilitate the in vivo validation of the mechanism of action (MOA) of new drugs. In high-content screening, by monitoring the real-time changes in omics data, AI technologies generalize these features and develop a model that can interpret the molecular and cellular MOA of new compounds and their associated pharmacokinetic, pharmacodynamic, toxicological, and bioavailability properties (Figure 4).
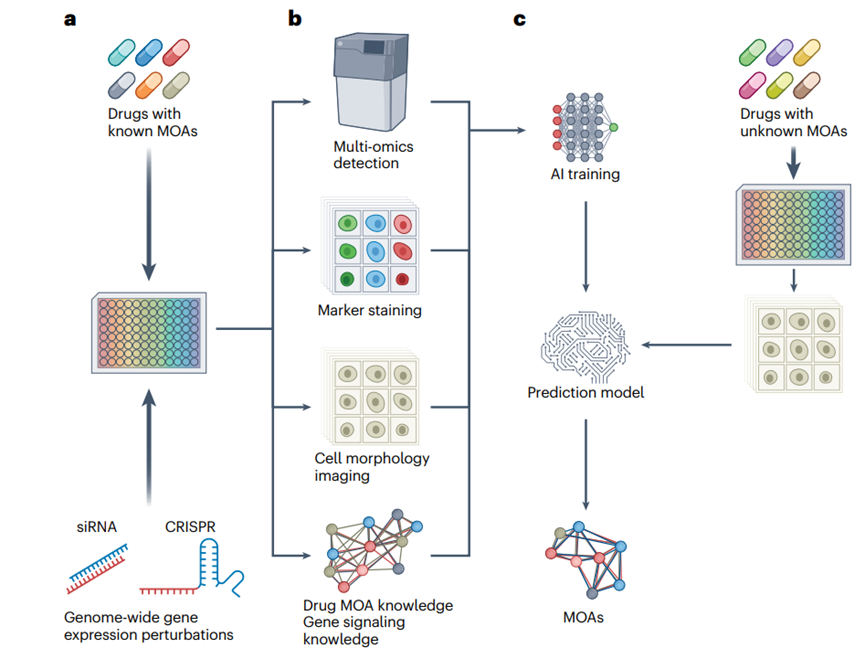
Figure 4. AI-driven MOA prediction
AI in Clinical Trials and the Real World
AI is increasingly guiding various aspects of clinical trials by analyzing patient data, including genetic information, clinical history, and lifestyle factors. Applying AI methods to such data can help identify biomarkers and patient characteristics that influence drug response, enabling more efficient and informative trial designs. By optimizing parameters such as patient selection, treatment regimens, and outcome measures, AI has the potential to increase trial success rates and accelerate the translation of drug candidates into clinical practice. Real-world data also provide a rich source of information from which AI applications can predict adverse events, drug-drug interactions, and other outcomes. The following sections describe key applications of AI in the clinical phase of drug development.
Biomarker Discovery
Biomarkers are biological indicators used to objectively measure and evaluate normal and pathological processes and treatment responses, and have great application value in the fields of medicine, biotechnology, and biopharmaceuticals. However, traditional hypothesis-driven biomarker discovery methods are often inefficient and fail to fully address the complexity of the disease. These methods are time-consuming and require a lot of resources for hypothesis validation, while the limitation of limited sample size hinders extensive validation in different populations.
Recent advances in artificial intelligence have greatly promoted biomarker discovery. Artificial intelligence models excel in identifying diagnostic biomarkers, providing predictive insights and diagnostic references for clinical pathology. A notable example is the “nuclei.io” digital pathology framework, which combines active learning with real-time human-computer interaction. This helps to efficiently build datasets and artificial intelligence models for various surgical pathology tasks by providing precise feedback to pathologists based on nuclear statistical data, thereby significantly improving diagnostic accuracy and efficiency.
Artificial intelligence also excels in identifying prognostic biomarkers that are critical for predicting disease progression and patient survival, thereby enabling targeted and personalized treatment. For example, deep learning models can characterize CD8+ T cell morphology in blood samples as an effective prognostic indicator for sepsis, distinguish nuclear features that mark cellular senescence, and identify proteomic biomarkers to accurately predict liver disease outcomes. AI can also predict prognostic biomarkers for various cancers, providing precise risk scores for survival, recurrence, and metastasis. Notably, survival analysis models using graph neural networks outperformed existing models and effectively distinguished risk groups beyond traditional clinical grading and staging—highlighting the potential of AI in prognostic enhancement and the critical collaboration between pathologists and AI.
In drug development, identifying predictive biomarkers is critical to improving research success by selecting patient populations most likely to benefit from treatment. These findings require rigorous prospective clinical validation. Although AI-based predictive biomarkers have not yet been applied clinically, proof-of-concept studies have shown that AI can predict patient response to treatment by predicting known biomarkers. The complexity of biological systems requires the integration of multiple types of biological data, including protein-protein interactions, into AI models for more comprehensive predictions.
Faced with the scarcity of large labeled datasets, researchers are deploying various strategies to optimize the application of AI in biomarker discovery. The integration of datasets from multiple sources shows great promise. Digital biomarkers from wearable sensors also expand the scope of discovery by providing rich longitudinal datasets. The identification of multimodal biomarkers through molecular diagnostics, radiomics, and histopathology imaging provides new avenues for precision medicine. In addition, population learning and automated dataset processing pipelines lay the foundation for large-scale, secure data collection.
However, AI models face challenges related to heterogeneity when applying their translational efficiency to clinical trials. Some studies have used deep learning to elucidate heterogeneity at the cellular and tissue levels and the diversity of tumor ecosystems, providing new avenues for disease subtype classification and patient stratification. Interpretability and trust are critical for the clinical acceptance of AI models and can be enhanced by integrating prior medical knowledge or embedding biological relationships into neural networks. Addressing bias issues in AI-driven biomarker discovery requires strategies such as validating models across geographically diverse patient cohorts and developing fair and transparent algorithms. Robust validation and responsible data management will facilitate the identification and application of biomarkers, supporting future drug development and disease treatment.
Predicting pharmacodynamic properties
The application of AI and big data tools can effectively solve pharmacodynamic problems and provide powerful tools for event time analysis, especially when dealing with high-dimensional data and nonlinear relationships in risk functions. AI supports personalized treatment by optimizing dose-response relationships, improving drug safety profiles, and optimizing therapeutic windows, which are all at the core of solving pharmacodynamic problems in precision medicine. Machine learning analysis of 442 small molecule kinases and 2,145 adverse events discovered new kinase-adverse event pairs, which can help risk mitigation and the development of safer small molecule kinase inhibitors. The Multi-omics Variational Autoencoder (MOVE) framework integrates multi-omics data to reveal drug interactions—such as the connection between metformin and the gut microbiota—and compares drug responses across various omics patterns. PharmBERT, a domain-specific language model, enhances drug safety by extracting key pharmacokinetic information from prescription labels, helping to identify adverse reactions and drug interactions. AI also optimizes drug dosage by analyzing genetic and physiological data, thereby providing personalized treatment recommendations for improved treatment outcomes. In addition, AI can analyze a patient’s genetic information, physiological characteristics, and past treatment responses to provide physicians with personalized dosage adjustment recommendations to optimize treatment outcomes.
Drug Repurposing
In addition to new drug discovery, AI is contributing to drug libraries by repurposing existing, approved drugs using large-scale biomedical datasets, thereby accelerating the development of optimal treatment options for various diseases. By discovering previously unrecognized therapeutic properties of approved drugs, AI reduces the time and costs associated with drug discovery. For example, AI has accelerated the repurposing of drugs for coronavirus disease 2019 (COVID-19), highlighting the value of AI in finding entirely new applications for existing drugs. AI can also facilitate drug repurposing by simulating clinical trials using real-world data, including electronic health records and insurance claims. For example, a deep learning recurrent neural network used causal reasoning and deep learning to analyze a medical claims database to effectively identify potential drug candidates. Applied to a cohort of millions of patients with coronary artery disease, it pinpointed drugs and combinations that enhanced treatment efficacy.
Another deep learning-based drug repurposing approach involves applying deep neural networks to omics data to classify drugs into therapeutic classes based on the transcriptional perturbations they induce in vitro. A study utilized perturbed samples from the LINCS project (https://lincsproject.org/) and 12 therapeutic categories from MeSH, achieving high classification accuracy—particularly on pathway-level data across a variety of biological systems and conditions—providing potential for drug repositioning. Feature attribution techniques were integrated with interpretable machine learning models to enhance the identification of gene expression signatures associated with synergistic drug responses. This strategy has been shown to improve feature interpretability and support the selection of optimal anticancer drug combinations based on molecular insights.
In addition, AI-based high-content screening can also be applied to drug repurposing (Figure 4). A deep learning model, MitoReID, was developed to identify mechanisms of action (MOA) through mitochondrial phenotyping. It provides a cost-effective, high-throughput solution for drug discovery and repurposing, validated with unseen drugs (not in the training set) and in vitro validation. By analyzing 570,096 cell images, MitoReID achieved 76.32% accuracy in identifying the mechanisms of action of drugs approved by the US Food and Drug Administration and successfully validated the cyclooxygenase-2 inhibition of epicatechin, a natural compound in tea. However, many of the challenges encountered in other stages of AI-driven drug development also apply to drug repurposing, including issues such as data quality, model interpretability, generalization ability, validation costs, regulatory barriers, integration with existing processes, and high computational requirements, which have hindered widespread adoption and practical applications.
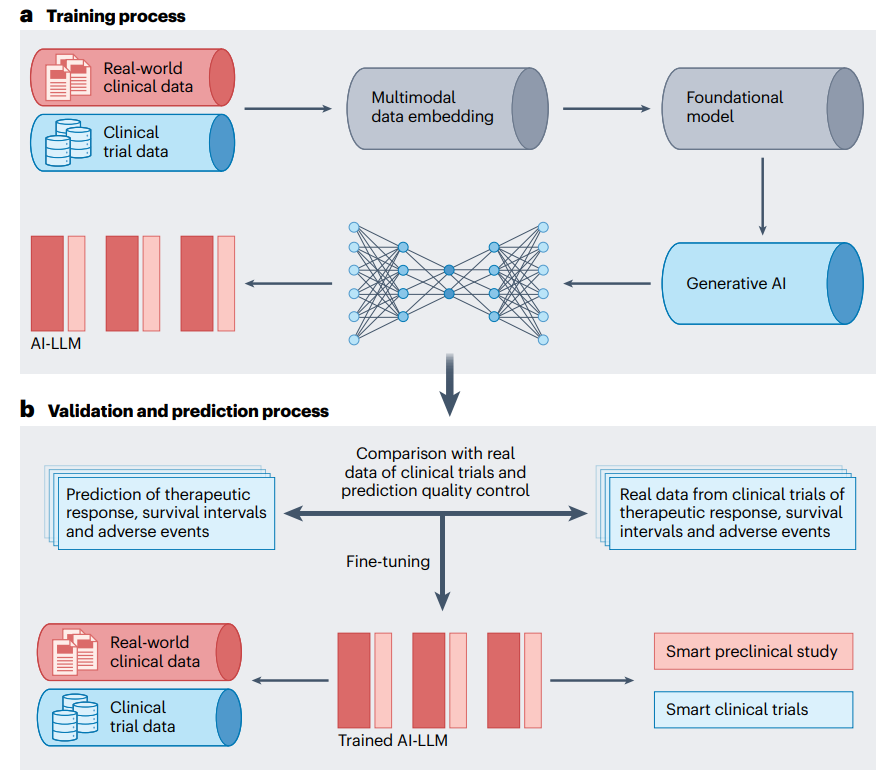
Figure 5. Application of AI in clinical practice
Improving trial efficiency and predicting outcomes
Clinical trials are often costly, time-consuming, and inefficient, with most trials facing delays in enrollment or difficulty finding enough volunteers. AI has the potential to optimize trial design, streamline recruitment, and predict patient responses, improving trial efficiency and success rates while reducing costs and time. An advanced process has been created that integrates multimodal datasets, uses AI to generate molecular lead compounds, ranks them by efficacy and safety, and uses deep reinforcement learning to create patentable analogs for testing. It also predicts Phase I/II clinical trial outcomes by estimating side effects and pathway activation, improving prediction accuracy and identifying potential risks in drug combinations. In real-world studies, AI can analyze data from electronic health records, insurance claims, and wearable devices to assess the effectiveness and safety of drugs (Figure 5). For example, a study using real-world data and the Trial Pathfinder tool simulated trial results from electronic health record data from 61,094 patients with advanced lung cancer, showing that relaxing trial criteria could double the number of eligible patients and improve survival outcomes. The approach has been validated in a variety of cancers, supporting more inclusive and safe trials.
Digital twin technology explored by Unlearn.ai can ease the challenge of finding suitable patients who meet inclusion criteria. The technology creates virtual copies of participants, allowing them to serve as control groups, thereby increasing the number of participants in experimental groups and improving trial efficiency. Unlearn.ai received a $12 million grant in April 2020 to advance the application, while other companies such as Novadiscovery and Jinkō are conducting digital twin-based clinical trial simulations for diseases such as lung cancer. The proposed approach uses computer modeling based on gene expression and clinical data, combined with deep learning and generative adversarial networks. By leveraging various health indicators, these digital twin technologies provide quantitative insights into important processes, provide dynamic health guidance and optimize treatment strategies. The approach aims to deepen the mathematical understanding of biological mechanisms, revolutionize clinical practice and fully personalize healthcare, for example, by generating patient-specific models that predict survival probability based on drug input. These models can also simulate clinical trials and optimize trial parameters to improve success rates. But they also have challenges, including high computational costs, complex process integration, ethical issues and limited personalization. These issues affect the accuracy of patient simulations, trial design and regulatory acceptance, thereby slowing innovation.
In addition to the clinical trial phase of drug development, AI can also analyze post-marketing surveillance data to support the safety, efficacy, and quality of drugs. The development and concurrent use of alternative approaches to identify and address safety issues early in the regulatory review process is critical to advancing regulatory science and optimizing drug development.
Challenges
Despite progress, no AI-developed drugs have yet entered Phase III clinical trials, highlighting the complexity of drug development. A key challenge is the lack of high-quality training data, which is attributed to high acquisition costs, privacy regulations, and limited data sharing—particularly for rare diseases or novel drug targets—which hinders the effectiveness of AI in identifying targets, biomarkers, and other features. In addition, existing data often contain missing information, errors, and biases, further reducing the reliability of AI. Drug discovery experiments can produce inconsistent results, and cost-saving measures may result in incomplete data. In addition, the underrepresentation of “negative” data (e.g., unsuccessful experiments and negative trial results) in the literature hinders a comprehensive understanding of drug-target-disease interactions, efficacy, and other clinical characteristics.
A key challenge in drug design is to strike a balance between multiple targets for success. Current studies often focus too much on chemical space and ignore other key factors (such as druggability and synthetic accessibility). Although multi-objective design methods are improving, developing effective scoring functions (e.g., affinity prediction and bioactivity) remains complex and requires extensive experiments. The lack of standardized evaluation processes further complicates model evaluation, especially when conflicting objectives arise, such as maximizing similarity to known bioactive molecules while achieving structural novelty. Despite the existence of benchmarking platforms such as MOSES and Guacamol, there is no consensus on best practices.
Appropriate molecular representation is key in generative models. Traditional methods such as SMILES and graphs are common and are being complemented by emerging data-driven methods such as self-supervised learning of hierarchical molecular graphs. However, capturing complexity and ensuring synthetic accessibility is difficult. Current methods for assessing synthetic feasibility are often imprecise, leading to the discovery of unsynthesizable molecules. Integrating reaction knowledge into molecule generation shows promise, but still needs improvement. Issues such as model interpretability, uncertainty in generating new molecules, and bias have become the focus of academic attention. Effectively integrating bias control with uncertainty estimation is essential to improve the quality of generated molecules.
Artificial intelligence faces challenges in so-called “undruggable” targets that lack suitable binding sites, including certain disordered proteins, transcription factors such as MYC and IRF4, and protein-protein interactions. New AI methods and high-content screening (Figure 4) that explore their conformational space and identify ligand binding sites may help overcome these obstacles.
Finally, technical challenges in algorithms and computing power limit the application of AI in drug development. Many AI algorithms used in drug development are designed for other fields and may not be fully applicable; for example, new algorithms based on natural language processing are needed to capture three-dimensional spatial interactions. In addition, the high computing resources required by AI methods pose a barrier, especially for small research teams. Collaborating with cloud providers and developing more efficient algorithms can help address these challenges. In addition, due to long cycles, low success rates, and uncertain returns, AI drug development faces talent shortages and investment risks, which affect investor confidence.
Future Directions
Artificial intelligence is revolutionizing the drug development process by extracting key insights from complex multi-omic biomedical data, identifying novel biomarkers, and detecting therapeutic targets and abnormalities, thereby facilitating the discovery of lead compounds and drug candidates. In addition, AI accelerates drug discovery, repurposing, and toxicity prediction, thereby reducing time, cost, and safety risks. However, the path to fully realize AI-driven advances in this field is still a work in progress, with many challenges to overcome and potential to be realized. Future efforts to address the above challenges should place particular emphasis on the following key directions.
First, developing new strategies to address the data scarcity problem in AI-driven drug development should be a top priority. Viable strategies to enhance data sharing, establish data standards, and develop new AI algorithms (e.g., “sparse” AI methods that can produce accurate predictions from very limited data) are essential. Multimodal pre-trained models that integrate textual and chemical information show promise in addressing the data scarcity problem, especially in zero-shot scenarios. By integrating a range of data such as genomics, transcriptomics, disease-specific molecular pathways, protein interactions, and clinical records, AI can also identify existing drugs with potential repurposing opportunities for overlooked or rare diseases.
Current approaches often focus on a single data type, thus missing the complex interrelationships between various biological systems. Establishing effective multimodal fusion methods can extract valuable insights from different sources and formats to advance drug development. With the rise of big data and GPU computing based on graphics processing units (GPUs, rather than traditional central processing units, CPUs), AI can now be applied to a variety of data forms, including text, images, and videos. Emerging models using omics data, including deep learning-based drug classification, have shown promise in drug efficacy prediction, mechanism identification, and toxicity assessment, highlighting the future potential of multimodal AI in drug development.
Many current AI models are purely data-driven, which limits their effectiveness in drug development due to the relative lack of sufficient high-quality data. Since our living systems all follow the principles of physics (also known as first principles), drugs are also subject to the constraints of physical laws without exception. Incorporating physical laws into existing data-driven AI algorithms is a future research direction that can help reduce data dependence and improve the accuracy and generalization ability of these models.
AI, especially large language models (LLMs), can ensure compliance with drug regulations by analyzing large amounts of documents and mastering the latest requirements. This improves efficiency, reduces the risk of noncompliance, and prevents delays in drug approvals. Developing AI models that are not only accurate but also explainable is critical to building trust between drug developers, regulators, clinicians, and patients by ensuring transparency and understanding of the decision-making process. These models can be incorporated early to optimize project funding and guide investments, thereby accelerating drug development.
The role of AI in medical modeling and simulation will be transformative in the coming decades. Advanced AI models will create increasingly detailed virtual human simulations, further enhancing our understanding of disease mechanisms, drug effects, and individual biological differences. Through simulation, AI can streamline clinical trial design and execution, testing different scenarios for optimal selection criteria to accelerate patient recruitment and improve the representativeness of trials. AI will also provide personalized medical decision support by analyzing health data and genomics, resulting in precise risk prediction, optimized treatment, and improved surgical guidance. Medical education will benefit from AI-driven virtual reality, providing more realistic training scenarios and improving the quality of medical services.
Conclusion
In summary, the continuous advancement of AI technology is significantly improving the efficiency and cost-effectiveness of drug development. However, it is important to recognize that AI is not a panacea. The advantage of AI technology lies in analyzing large and complex data and assisting rapid decision-making to supplement human functions and enhance human capabilities, but AI is not intended to completely replace human ingenuity or authority. AI-designed drugs and predicted properties still need to be verified through wet lab experiments, and human input is still needed to determine the direction of AI research and use. However, given the growing capabilities and pace of progress in AI, as well as the open source of large models including the recent AlphaFold3, we can be cautiously optimistic about the prospects of AI in accelerating drug development and benefiting human health.
Reference
Zhang K, Yang X, Wang Y, Yu Y, Huang N, Li G, Li X, Wu JC, Yang S. Artificial intelligence in drug development. Nat Med. 2025 Jan;31(1):45-59. doi: 10.1038/s41591-024-03434-4. Epub 2025 Jan 20. PMID: 39833407.