Mitapivat: A Novel Treatment for Pyruvate Kinase Deficiency and Hereditary Hemolytic Anemias
Abstract
Mitapivat is a first-in-class, oral, and selective activator of pyruvate kinase-R (PKR) enzymes, which play a crucial role in the production of red blood cells. Mitapivat is being investigated for the treatment of various hemolytic anemias, including pyruvate kinase deficiency (PKD), a rare genetic disorder that causes the destruction of red blood cells. By activating PKR enzymes, Mitapivat increases the production of ATP, a molecule that provides energy to red blood cells and helps them survive. Clinical studies have shown promising results with Mitapivat in patients with PKD, improving their anemia and reducing their need for blood transfusions.
Hereditary hemolytic anemias and Pyruvate Kinase Deficiency
Hereditary hemolytic anemias (HHA) are a group of inherited disorders characterized by the destruction of red blood cells, resulting in chronic anemia. There are many types of HHA, each with its specific genetic mutations, pathophysiology, and clinical presentation. These disorders can range from mild to severe and may be asymptomatic or cause significant morbidity and mortality. Pyruvate Kinase Deficiency (PKD) is one of the most common types of HHA, caused by mutations in the PKLR gene, which encodes the enzyme pyruvate kinase. PKD affects the metabolic pathway of red blood cells, leading to their premature destruction.
PKD is an autosomal recessive disorder, meaning that individuals need to inherit two defective copies of the PKLR gene, one from each parent, to develop the disease. The prevalence of PKD varies by ethnic group and ranges from 1 in 20,000 in Northern Europe to 1 in 200,000 in other populations. PKD usually presents in childhood or early adulthood, although milder forms of the disease can manifest later in life. Patients with PKD often have chronic hemolytic anemia, which can lead to fatigue, pallor, jaundice, and splenomegaly. Additionally, patients with PKD may develop complications such as gallstones, leg ulcers, and pulmonary hypertension.
Treatment of PKD focuses on managing the anemia and preventing complications. Blood transfusions are often required in severe cases, but they can lead to iron overload, which requires chelation therapy. Splenectomy is another treatment option, which can improve anemia but increases the risk of infections. Emerging therapies such as Mitapivat, a pyruvate kinase activator, have shown promising results in clinical trials, reducing the need for blood transfusions and improving anemia in patients with PKD.
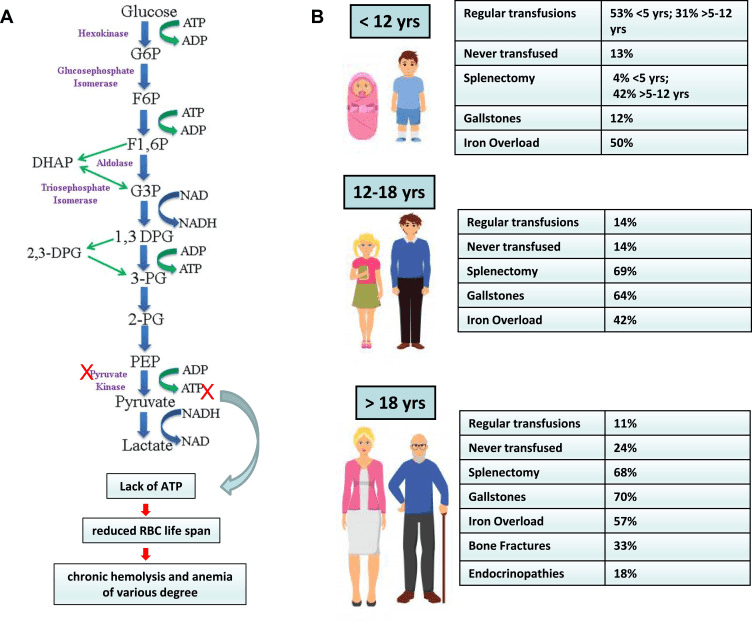
Fig 1. The physiopathology of pyruvate kinase deficiency (PKD), and its clinical features and complications. (A) The Embden-Meyerhof pathway alteration. (B) Different clinical features and complications during infancy, adolescence, and adulthood [1].
Mitapivat and its mechanism of action
Mitapivat is a novel pyruvate kinase activator that has shown promising results in the treatment of hereditary hemolytic anemias (HHA). It works by increasing the activity of pyruvate kinase, an enzyme that plays a crucial role in the metabolic pathway of red blood cells. In patients with HHA, mutations in the genes that code for enzymes involved in this pathway lead to the premature destruction of red blood cells. By activating pyruvate kinase, Mitapivat helps to increase the production of ATP and reduce the accumulation of 2,3-DPG, which can improve the survival of red blood cells. This mechanism of action has been demonstrated in preclinical studies and clinical trials, showing significant reductions in hemolysis and improvements in anemia.
Mitapivat pharmacology
Mitapivat as a novel small molecule drug that has shown promising results in the treatment of hereditary hemolytic anemias (HHA). It is a pyruvate kinase activator that increases the activity of pyruvate kinase, an enzyme that plays a crucial role in the metabolic pathway of red blood cells. The pharmacology of Mitapivat involves the role of PKR enzyme in red blood cell metabolism, the mechanism of action of Mitapivat in PKR activation, and its pharmacokinetics and pharmacodynamics.
PKR enzyme and its role in red blood cell metabolism
Pyruvate kinase (PK) is an enzyme that catalyzes the conversion of phosphoenolpyruvate to pyruvate, a key step in the glycolytic pathway that generates ATP. PKR is the red blood cell-specific isoform of PK that is essential for the survival and function of red blood cells. Mutations in the PKR gene lead to reduced PKR activity, which results in decreased ATP production and increased levels of 2,3-DPG, a molecule that reduces the affinity of hemoglobin for oxygen.
Mechanism of action of Mitapivat in PKR activation
Mitapivat binds to the allosteric site of PKR and stabilizes the R-state conformation, leading to an increase in PKR activity. This results in increased ATP production and decreased levels of 2,3-DPG, which improve the survival and function of red blood cells. Mitapivat has been shown to be effective in reducing hemolysis and improving anemia in preclinical studies and clinical trials.
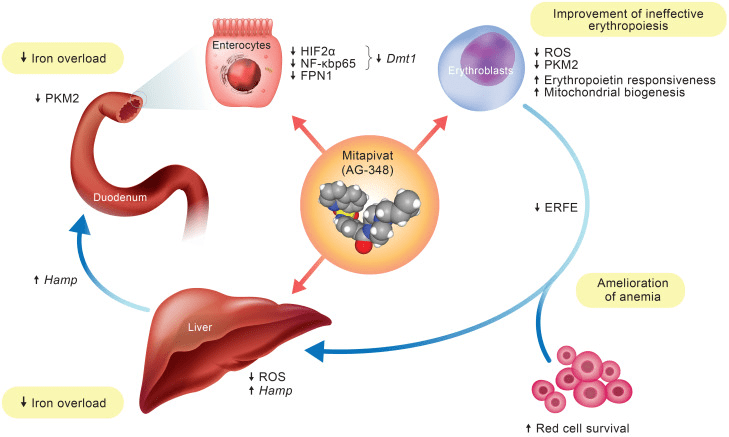
Fig 2. Schematic diagram of mitapivat actions in β-thalassemic mouse model [2].
Pharmacokinetics and pharmacodynamics of Mitapivat
Mitapivat is administered orally, and it has a high oral bioavailability. It reaches peak plasma concentration within 2 to 3 hours of administration and has a half-life of approximately 24 hours. Mitapivat is primarily metabolized by the liver, and its metabolites are excreted in the urine and feces. The pharmacodynamics of Mitapivat has been studied in preclinical and clinical studies, which have shown that it increases PKR activity, reduces hemolysis, and improves anemia in patients with HHA.
In conclusion, Mitapivat is a promising drug for the treatment of hereditary hemolytic anemias. Its mechanism of action involves activating the PKR enzyme, which plays a crucial role in red blood cell metabolism. Mitapivat has a favorable pharmacokinetic profile, and its pharmacodynamics has been extensively studied, showing significant reductions in hemolysis and improvements in anemia.
Clinical studies with Mitapivat
Mitapivat has been studied in several clinical trials for the treatment of hereditary hemolytic anemias, including Pyruvate kinase deficiency (PKD) and Non-spherocytic hemolytic anemia (NSHA).
In a Phase 2 study involving 52 patients with PKD, Mitapivat demonstrated significant improvements in hemoglobin levels and reduced transfusion requirements. The study showed that Mitapivat treatment resulted in a mean increase in hemoglobin levels of 1.6 g/dL from baseline, with 45% of patients achieving a hemoglobin increase of at least 1.5 g/dL. The study also showed that 41% of patients achieved transfusion independence during the treatment period.
In another Phase 2 study involving 24 patients with NSHA, Mitapivat demonstrated a reduction in hemolysis and an increase in hemoglobin levels. The study showed that treatment with Mitapivat resulted in a mean reduction in bilirubin levels of 38% and a mean increase in hemoglobin levels of 1.1 g/dL from baseline.
Overall, these studies suggest that Mitapivat has the potential to be an effective treatment option for patients with hereditary hemolytic anemias, including PKD and NSHA.
Table 1. Currently ongoing and planned clinical trials evaluating mitapivat for the treatment of hereditary hemolytic anemias [3].

Future directions and conclusion
Mitapivat, a novel pyruvate kinase activator, has shown promising results in the treatment of hereditary hemolytic anemias such as PKD and NSHA. However, further research is required to explore the potential applications of Mitapivat in other hemolytic anemias. The drug has the potential to be effective in the treatment of other red blood cell disorders, such as sickle cell disease, thalassemia, and other types of hemolytic anemias.
Future clinical trials will likely focus on exploring the long-term safety and efficacy of Mitapivat in larger patient populations. Additionally, further research is needed to investigate the optimal dosing and treatment duration of Mitapivat for the treatment of hereditary hemolytic anemias.
The promising results of the Phase 2 studies suggest that Mitapivat may be a valuable addition to the current treatment options for hereditary hemolytic anemias. Mitapivat has shown the potential to improve anemia and reduce the need for blood transfusions in patients with PKD and NSHA. Its mechanism of action, which involves activating the PKR enzyme in red blood cells, makes it a promising candidate for the treatment of other hemolytic anemias.
In conclusion, Mitapivat is a promising new treatment option for hereditary hemolytic anemias. Its ability to activate the PKR enzyme and reduce hemolysis has shown significant improvements in hemoglobin levels and reduced transfusion requirements. With further research and clinical trials, Mitapivat has the potential to become a valuable treatment option for patients with hereditary hemolytic anemias and may also have applications in the treatment of other red blood cell disorders.
Reference
- Fattizzo, B., Cavallaro, F., Marcello, A. P. M. L., Vercellati, C., & Barcellini, W. (2022). Pyruvate Kinase Deficiency: Current Challenges and Future Prospects. Journal of blood medicine, 13, 461–471.
- Matte, A., Federti, E., Kung, C., Kosinski, P. A., Narayanaswamy, R., Russo, R., Federico, G., Carlomagno, F., Desbats, M. A., Salviati, L., Leboeuf, C., Valenti, M. T., Turrini, F., Janin, A., Yu, S., Beneduce, E., Ronseaux, S., Iatcenko, I., Dang, L., Ganz, T., … De Franceschi, L. (2021). The pyruvate kinase activator mitapivat reduces hemolysis and improves anemia in a β-thalassemia mouse model. The Journal of clinical investigation, 131(10), e144206.
- Al-Samkari, H., & van Beers, E. J. (2021). Mitapivat, a novel pyruvate kinase activator, for the treatment of hereditary hemolytic anemias. Therapeutic advances in hematology, 12, 20406207211066070.
- Luke, N., Hillier, K., Al-Samkari, H., & Grace, R. F. (2023). Updates and advances in pyruvate kinase deficiency. Trends in molecular medicine, 29(5), 406–418.
- Song, A. B., & Al-Samkari, H. (2022). An evaluation of mitapivat for the treatment of hemolytic anemia in adults with pyruvate kinase deficiency. Expert review of hematology, 15(10), 875–885.
- Benedetto Tiz, D., Bagnoli, L., Rosati, O., Marini, F., Santi, C., & Sancineto, L. (2022). FDA-Approved Small Molecules in 2022: Clinical Uses and Their Synthesis. Pharmaceutics, 14(11), 2538.