Targeted Protein Degradation and PROTACs: Transforming the Future of Cancer Therapy
Abstract
Targeted Protein Degradation (TPD) is revolutionizing cancer treatment by offering a novel mechanism to eliminate disease-driving proteins rather than merely inhibiting them. At the forefront of this innovation are PROTACs (Proteolysis-Targeting Chimeras), which harness the cell’s own degradation machinery to selectively remove oncogenic proteins. This blog explores the fundamental principles behind PROTACs, highlights their clinical applications in cancers like prostate and breast cancer, and examines the limitations that researchers are actively working to overcome. It also introduces next-generation degradation platforms, including LYTACs, AUTACs, and molecular glues, that extend the reach of TPD beyond intracellular targets. Together, these advancements signify a new era in precision oncology, expanding therapeutic possibilities and addressing previously undruggable targets.
Why Protein Degradation Matters in Cancer Therapy
For decades, cancer drug development has centered on inhibiting the activity of specific proteins—especially enzymes like kinases—that drive tumor growth. While many small-molecule inhibitors (SMIs) and monoclonal antibodies have revolutionized treatment, they come with significant limitations. Chief among them: they can only target proteins that have an accessible active site or surface pocket. This leaves roughly 85% of the human proteome—including transcription factors, scaffold proteins, and other non-enzymatic molecules—undruggable by conventional means.
That’s where Targeted Protein Degradation (TPD) enters the stage.
Rather than blocking a protein’s function, TPD eliminates the protein altogether by hijacking the cell’s natural protein disposal system. This is especially important in oncology, where rogue proteins—often mutated or overexpressed—can evade inhibition but remain vulnerable to degradation.
The most well-known tool in the TPD toolbox is the Proteolysis-Targeting Chimera (PROTAC). These small, bifunctional molecules bind both the protein of interest and an E3 ubiquitin ligase, bringing them into proximity. The result? The protein is tagged for degradation by the ubiquitin-proteasome system (UPS) and destroyed by the cell’s proteasome.
This degradation-first approach brings several benefits:
It can target previously “undruggable” proteins.
It reduces the likelihood of drug resistance due to transient binding.
It is event-driven, meaning one PROTAC molecule can trigger multiple degradation cycles.
With the first PROTAC drugs now in clinical trials for cancers like prostate, breast, and blood malignancies, TPD is rapidly becoming a cornerstone of next-generation oncology therapies.
What Are PROTACs and How Do They Work?
PROTACs (Proteolysis-Targeting Chimeras) represent a transformative class of molecules that operate by a fundamentally different mechanism than traditional drugs. Instead of simply blocking the function of a disease-causing protein, PROTACs induce its degradation, effectively removing it from the cell.
A PROTAC is a heterobifunctional molecule composed of three parts:
A warhead that binds to the protein of interest (POI).
A ligand that binds to an E3 ubiquitin ligase, a component of the cell’s protein degradation machinery.
A linker that connects the two.
The key to PROTAC technology is the formation of a ternary complex: the PROTAC simultaneously binds both the POI and the E3 ligase, bringing them into close proximity. This allows the E3 ligase to tag the POI with ubiquitin molecules, marking it for destruction by the 26S proteasome—the cell’s “garbage disposal” system.
One of the most significant advantages of PROTACs is their event-driven action. Unlike traditional inhibitors that must remain bound to their targets to be effective, PROTACs can act catalytically—after initiating the degradation of one protein, they are free to engage others, amplifying their therapeutic effect.
Another benefit is their ability to target “undruggable” proteins—those without enzymatic activity or with flat, featureless surfaces, such as transcription factors. By leveraging the cellular degradation pathway, PROTACs can bypass the need for classical binding pockets.
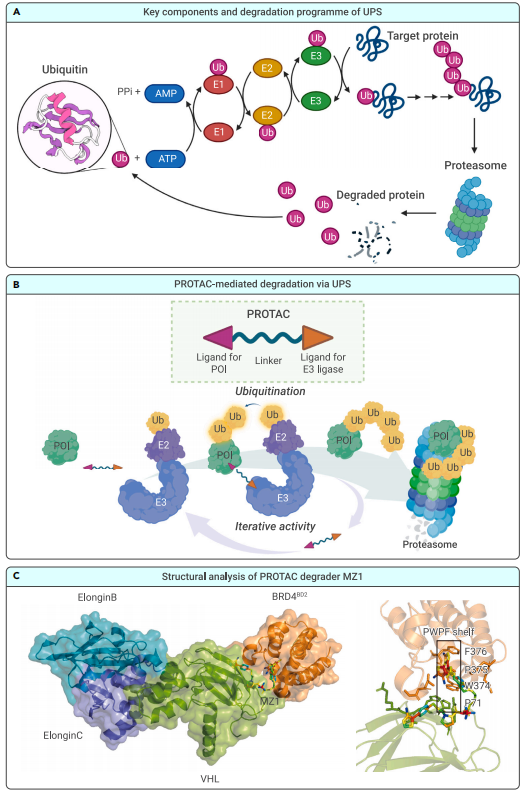
Figure 1. Background information on PROTACs
Structural studies, such as the crystal structure of the BRD4-MZ1-VHL ternary complex, have revealed the importance of protein–protein interactions and linker geometry in promoting stable and selective degradation. These insights are helping to refine PROTAC design for greater potency and specificity.
As research progresses, PROTACs are not only emerging as powerful cancer therapies but also as tools for dissecting complex biological systems.
Applications in Cancer — From Theory to Clinical Trials
The therapeutic promise of PROTACs (Proteolysis-Targeting Chimeras) has moved beyond theory into the realm of clinical oncology, where these innovative molecules are being tested in humans for the treatment of various cancers. Unlike traditional inhibitors that often face resistance or limited applicability, PROTACs offer a more robust strategy by targeting disease-related proteins for complete degradation.
In the clinical pipeline, several PROTACs have shown encouraging results, particularly in hormone-driven cancers. One standout example is ARV-110 (bavdegalutamide), developed by Arvinas, which targets the androgen receptor (AR) in metastatic castration-resistant prostate cancer (mCRPC). ARV-110 has demonstrated the ability to degrade both wild-type and mutant AR, leading to significant prostate-specific antigen (PSA) reductions in patients, and offering a new approach where traditional antiandrogens have failed.
Similarly, ARV-471 targets the estrogen receptor (ER) in breast cancer and is currently undergoing Phase II trials. It has been well-tolerated and has shown early signs of clinical benefit, including tumor shrinkage in patients with advanced ER-positive, HER2-negative breast cancer. The success of these hormone receptor-targeting PROTACs signals a paradigm shift in how endocrine-driven malignancies may be managed in the future.
Beyond hormone receptors, PROTACs are also being developed against other oncogenic proteins such as BTK (Bruton’s tyrosine kinase), BCL-xL, STAT3, and BRD9. Many of these targets are implicated in hematological malignancies like B-cell lymphomas and acute leukemias, where drug resistance and relapse remain significant challenges.
With over 20 PROTAC candidates currently in clinical or preclinical development, the field is rapidly evolving. These drugs are not only reshaping how we target “undruggable” proteins but also offering precision medicine options that can be tailored to individual cancer profiles.
Challenges and Limitations in PROTAC Development
While PROTACs (Proteolysis-Targeting Chimeras) represent a major advancement in targeted cancer therapy, their development and clinical application are not without hurdles. As these molecules transition from laboratory research to clinical settings, several key challenges and limitations have emerged that must be addressed to fully harness their therapeutic potential.
One major obstacle is the “hook effect,” a phenomenon where excessively high concentrations of a PROTAC can paradoxically reduce degradation efficiency. This happens when PROTAC molecules saturate both the target protein and the E3 ligase independently, preventing the formation of the productive ternary complex necessary for ubiquitination. This effect complicates dose optimization in both preclinical and clinical studies.
Another significant limitation is poor cell permeability and bioavailability. PROTACs are typically larger and more complex than traditional small-molecule drugs, often exceeding Lipinski’s “Rule of 5” parameters. Their size and polarity can hinder cellular uptake, limit tissue penetration, and reduce oral bioavailability—posing formulation and delivery challenges.
Moreover, the limited diversity of usable E3 ligases is a bottleneck in PROTAC design. Most current PROTACs recruit either cereblon (CRBN) or von Hippel–Lindau (VHL) ligases. However, these ligases are not universally expressed across all tissues or tumor types, which restricts the range of applicable targets and may cause off-target toxicity in healthy cells that also express the chosen E3 ligase.
Finally, toxicity and selectivity issues remain an area of concern. Degrading a protein rather than inhibiting it can lead to permanent loss of function, which may be harmful if the protein has essential roles in normal tissues. Additionally, off-target degradation due to unintended protein-protein interactions or poor selectivity can contribute to adverse effects.
To overcome these limitations, researchers are exploring novel E3 ligases, improving linker chemistry, and developing controlled-release systems to improve PROTAC pharmacokinetics and therapeutic windows.
Beyond PROTACs — The Expanding TPD Universe
While PROTACs (Proteolysis-Targeting Chimeras) have garnered significant attention for their ability to degrade intracellular proteins, the Targeted Protein Degradation (TPD) landscape is rapidly expanding with next-generation technologies that go far beyond the classical PROTAC framework. These innovative approaches aim to overcome limitations in cellular permeability, tissue specificity, and the ability to target extracellular or membrane-bound proteins.
One such advancement is the development of LYTACs (Lysosome-Targeting Chimeras). Unlike PROTACs, which rely on the ubiquitin-proteasome system, LYTACs harness the lysosomal degradation pathway to eliminate extracellular or membrane-associated proteins. They achieve this by binding the target protein and delivering it to the lysosome via receptors like CI-M6PR or ASGPR. This platform opens up the possibility of degrading secreted factors and immune checkpoint proteins—targets historically inaccessible to PROTACs.
Another class of degraders includes AUTACs (Autophagy-Targeting Chimeras) and ATTECs (Autophagosome-Tethering Compounds), which exploit the autophagy pathway. These molecules label disease-causing proteins or even organelles (like damaged mitochondria) for autophagic degradation. This strategy is particularly valuable in neurodegenerative diseases and certain cancers where dysfunctional proteins accumulate in the cytosol.
The rise of molecular glues—smaller, simpler molecules that stabilize interactions between a target protein and an E3 ligase—also represents a promising frontier. Unlike bifunctional PROTACs, molecular glues operate through allosteric modulation, enhancing the ubiquitination of target proteins without the need for a linker. Several molecular glues, such as lenalidomide and pomalidomide, are already approved for clinical use in hematological malignancies.
Finally, researchers are exploring photoactivatable PROTACs, dual-targeting (trivalent) PROTACs, and CHAMPs (chaperone-mediated protein degraders) to further refine degradation precision, spatiotemporal control, and target range.
Collectively, these innovations are broadening the therapeutic horizons of TPD, making it a versatile and customizable approach to treat a wide range of diseases—including cancers, autoimmune disorders, and neurodegeneration.
References
Békés, M., Langley, D. R., & Crews, C. M. (2022). PROTAC targeted protein degraders: The past is prologue. Nature Reviews Drug Discovery, 21(3), 181–200.
https://doi.org/10.1038/s41573-021-00372-7
Li, J., Chen, X., Lu, A., & Liang, C. (2023). Targeted protein degradation in cancers: Orthodox PROTACs and beyond. The Innovation, 4(3), 100413.
https://doi.org/10.1016/j.xinn.2023.100413
Bondeson, D. P., & Crews, C. M. (2017). Targeted protein degradation by small molecules. Annual Review of Pharmacology and Toxicology, 57, 107–123.
https://doi.org/10.1146/annurev-pharmtox-010716-104952
Churcher, I. (2018). Protac-induced protein degradation in drug discovery: Breaking the rules or just making new ones? Journal of Medicinal Chemistry, 61(2), 444–452.
https://doi.org/10.1021/acs.jmedchem.7b01275
Gadd, M. S., Testa, A., Lucas, X., et al. (2017). Structural basis of PROTAC cooperative recognition for selective protein degradation. Nature Chemical Biology, 13(5), 514–521.
https://doi.org/10.1038/nchembio.2329
Sakamoto, K. M., Kim, K. B., Kumagai, A., et al. (2001). Protacs: Chimeric molecules that target proteins to the SCF complex for ubiquitination and degradation. Proceedings of the National Academy of Sciences, 98(15), 8554–8559.
https://doi.org/10.1073/pnas.141230798
Bondeson, D. P., Mares, A., Smith, I. E. D., et al. (2015). Catalytic in vivo protein knockdown by small-molecule PROTACs. Nature Chemical Biology, 11(8), 611–617.
https://doi.org/10.1038/nchembio.1858
Gao, X., Burris, H. A., Vuky, J., et al. (2022). Phase 1/2 study of ARV-110, an androgen receptor (AR) PROTAC degrader, in metastatic castration-resistant prostate cancer (mCRPC). Journal of Clinical Oncology, 40(16_suppl), 17.
https://doi.org/10.1200/JCO.2022.40.16_suppl.17
Pettersson, M., & Crews, C. M. (2019). PROteolysis TArgeting Chimeras (PROTACs)—Past, present and future. Drug Discovery Today: Technologies, 31, 15–27.
https://doi.org/10.1016/j.ddtec.2019.01.002
Banik, S. M., Pedram, K., Wisnovsky, S., et al. (2020). Lysosome-targeting chimaeras for degradation of extracellular proteins. Nature, 584(7820), 291–297.