The Hidden Meaning of “New Uses for Old Drugs”
Abstract
Discovering new drugs is a labor-intensive, costly, and time-consuming endeavor. De novo design introduces innovative approaches to treatment. However, developing new drugs from scratch typically involves significant effort and has a low success rate. An alternative strategy is to utilize approved drugs for new drug development, which can be an efficient and practical approach.
Using approved drugs for new drug development goes beyond simply finding “new uses for old drugs.” The “new uses for old drugs” strategy involves repositioning existing medications to uncover new applications and treating diseases that differ from their original approved uses.
Additionally, another approach to leveraging approved drugs is to modify the molecular structure of these existing medications and explore the feasibility of chemical synthesis methods. However, resources can sometimes be limited. The key to effectively utilizing new drug development based on approved drugs lies in maximizing the potential of chemical modifications and computer-assisted techniques.
The road to new drug development is long and arduous
New drug development is a long and arduous project. Many projects require billions of dollars of investment and last for more than 10 years. Among them, the early R&D stage, from the discovery of hit compounds to the optimization of lead compounds, accounts for about 20% of the total budget.
For small molecule drug development, this budget is mainly used for the chemical synthesis and pharmacological evaluation of thousands of similar derivatives. Often, only one out of every 5,000-10,000 new molecular entities is finally approved.
In this exploration process, there are many strategies available, such as structure-assisted drug design, natural product-based methods, high-throughput hit discovery methods, etc., each with its own advantages. And one research strategy is based on drugs that have already been on the market, using this as a privileged starting point for re-research and development.
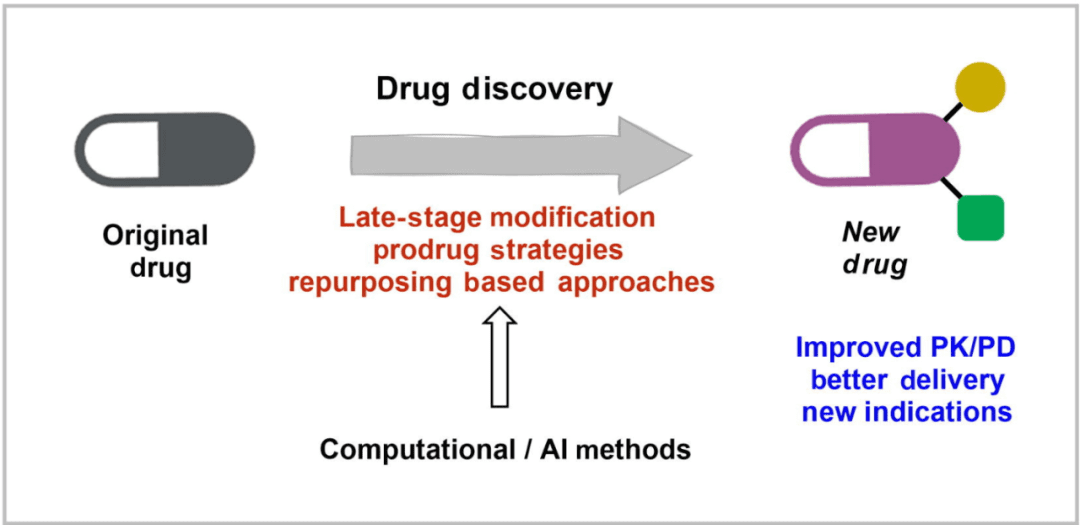
As shown in the figure above, the development of new drugs based on marketed drugs mainly includes structural transformation, prodrug strategy, and reuse as the starting point for discovering new biologically active molecular entities (i.e., new uses of old drugs). In these processes, computer/artificial intelligence-based methods are indispensable.
Moreover, this R&D strategy is not limited to “me-too” drugs, but also includes “me-better” and first-in-class innovative drugs.
New drug development strategy based on approved drugs
2.1. Chemical transformation of original drugs
Late functionalization and molecular editing
Late functionalization (LSF) is a chemical transformation measure that introduces substituents into complex molecular structures through chemical, regional, or stereoselective selection.
It is usually through the direct functionalization of the original drug structure through unactivated C-H bonds, generally involving mild conditions and diverse procedures, such as photoredox, free radical and transition metal chemistry, electrochemistry, etc.
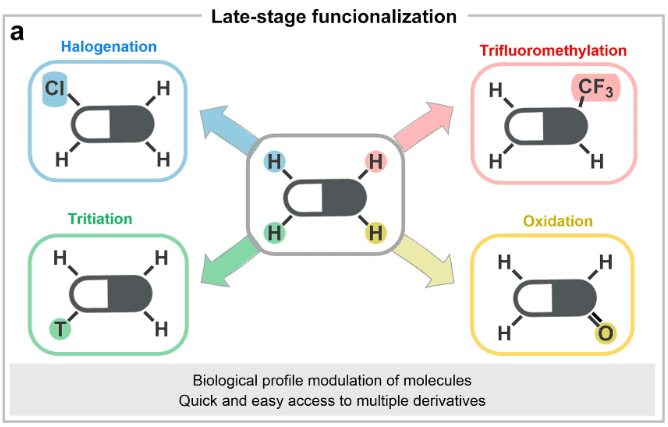
At present, feasible late functional modifications of related drugs include halogenation, methylation, trifluoromethylation, tritiation (for metabolic tracking research), oxidation, amination, borylation, acylation, etc., as shown above.
The goal of late functional modification in drug discovery is to explore how structural modification of drugs can improve their biological and physicochemical properties.
In addition, chemical transformation strategies that can be used to achieve the above goals include late skeleton editing technology, including: expansion, contraction, single atom exchange, molecular isomerization, and functional group migration of heteroaromatic hydrocarbon rings.
Late functional modification is widely used in current medicinal chemistry, avoiding the tedious work of designing and synthesizing each individual compound from scratch in the optimization of lead compounds. It plays the greatest role by combining with rational drug design and computer tools. Moreover, this efficient strategy is not only applicable to small molecule organic compounds, but also to the preparation of highly complex compound molecules for peptide and natural product synthesis and derivatization, as well as protein functionalization.
Case Study 1: Loratadine
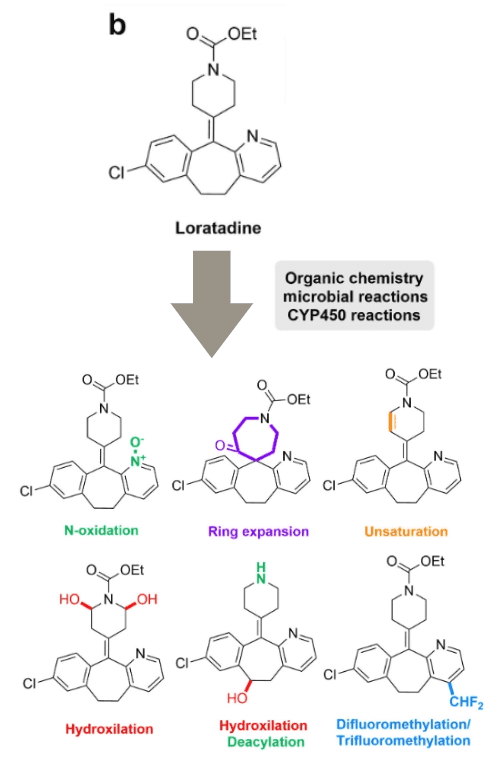
Loratadine is a common antihistamine. Pfizer has explored the chemical space around it and used some chemical transformation methods for late functional modification, including electrochemical and photochemical reactions, Minisci reactions, and biochemical processes such as microsomal CYP450 hydroxylation or microbial deacylation. As shown in the figure above, these methods ultimately improved the drug efficacy and metabolic stability of the original drug.
Case Study 2: Platinum
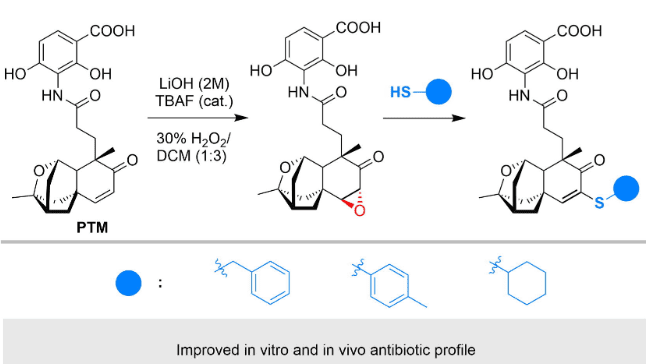
Platinum thiabendazole (PTM) is a naturally occurring antibiotic. As shown in the figure above, by installing an epoxide on its structure, the original drug has better antibiotic activity in vitro and greatly improves the survival rate of in vivo models. The modified structure has not yet been approved for marketing, but its changes in efficacy and properties illustrate the advantages of late-stage modification in quickly obtaining more potent lead compounds for complex molecules.
Molecular editing is a programmatic modification of a single structural pattern (atom, chain, or ring) in the parent molecular structure, including contraction and expansion. Different N-heterocyclic skeletons, in a given target binding, may sometimes only change one atom pair, which will produce huge differences in biological activity or biological application.
Heterocyclic skeletons are very common in many approved drugs (see previous reviews at the end of the article) and diverse. From the perspective of cost and time efficiency, the use of molecular editing to transform drug heterocyclic skeletons has far-reaching significance in future drug discovery.
In rational design, adjusting the structure and chemical properties to determine the small molecule synthesis method is crucial for the development of candidate drugs. In the later stage of functional modification, challenging scaffolds containing several carbon-hydrogen bonds with different physicochemical properties may be involved, which are sometimes difficult to break through by human methods.
Computational science comes to the rescue!
It has been reported that a computational science-based deep learning (DL) method, specifically graph neural networks (GNNs), was used to automatically screen C-H borylation reactions through the late functionalization modification process, and finally identify the impact on late hits.
In addition, the researchers also used graph characterization and other quantum mechanical techniques, such as density functional theory (DFT), to improve the prediction of regioselectivity. Therefore, a large amount of data is provided for DL to identify suitable drugs that can be derived through late functionalization modification. Even the optimal reaction conditions analysis for subsequent high-throughput experiment (HTE) screening can be performed.
Moreover, GNNs trained on 3D graphs have better prediction performance than GNNs trained on 2D graphs. The former can provide relevant information about the geometric atomic environment and show higher regioselectivity of chemical reactions.
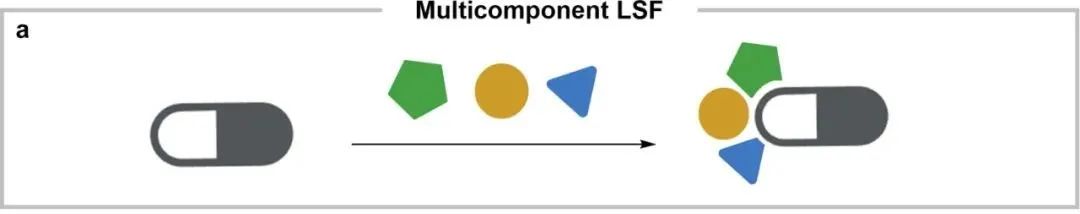
Multicomponent reaction: The original drug is used as one of the substrates in a multicomponent reaction, which is similar to the functional modification in the later stage, but will cause drastic changes in the core of the drug. The conceptual diagram is shown in Figure a below.
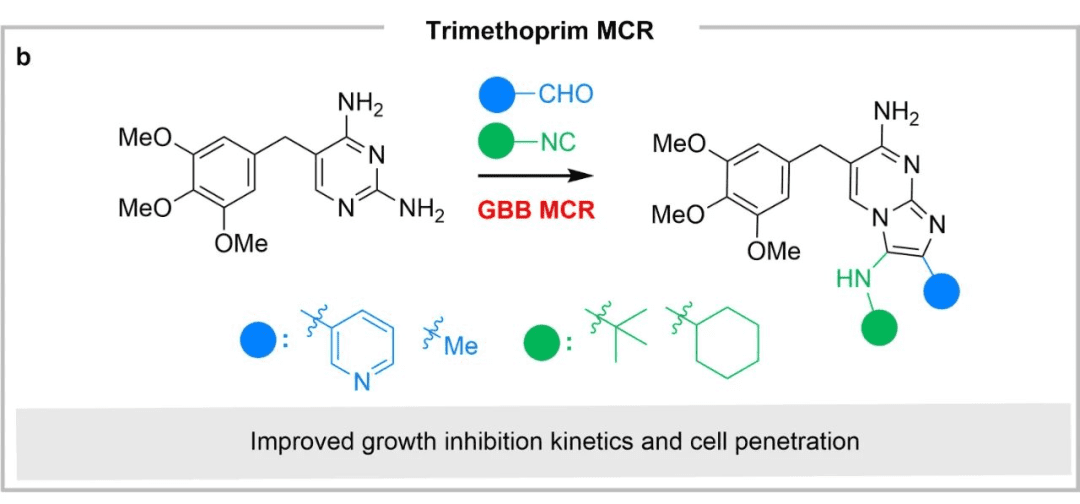
Case Study 1: Trimethoprim
Trimethoprim is an important antibiotic. The pyrimidine core was modified into an imidazopyridine core through a GBB multicomponent reaction. The new derivatives showed better growth inhibition kinetics and cell permeability while maintaining the original antibiotic activity.
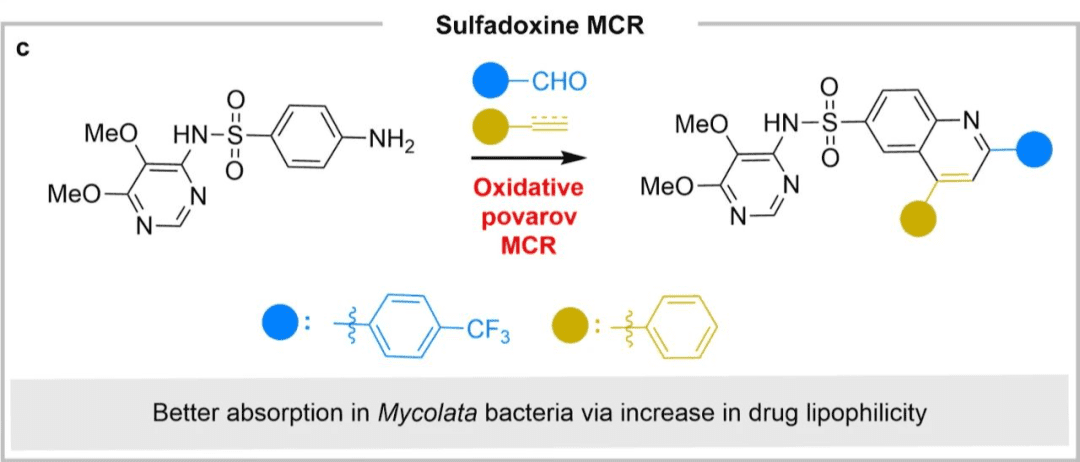
Case Study 2: Antiprotozoal Sulfadoxine
Another example of multi-component late-stage modification is shown in the figure below. Through the oxidative Povarov multi-component reaction, the antiprotozoan sulfadoxine was converted into an antifungal agent (the bacterial group to which tuberculosis belongs), and its aniline motif was converted into a substituted quinoline, thereby increasing its lipophilicity and enabling better absorption of fungi and bacteria.
2.2. “Prodrug” strategy
Prodrugs are generally inactive compounds that need to be metabolized to provide the desired biological activity and ideal therapeutic effect. The prodrug strategy is mainly to overcome the toxicity and PK issues that often affect drug performance.
The concept of prodrugs began in 1958 and currently accounts for about 10% of the total marketed drugs, fully demonstrating the impact of biotransformation on the drug discovery paradigm.
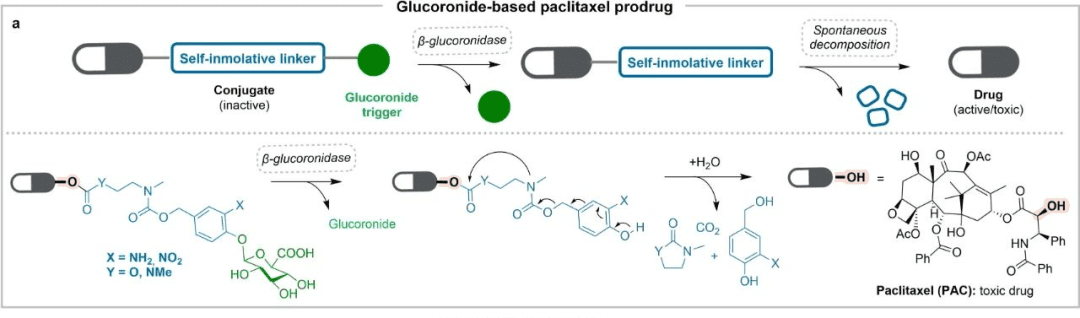
The most typical example is paclitaxel (pictured above), which is also a milestone in cancer treatment. Severe solubility problems, multidrug resistance problems, toxicity problems, etc. severely limit the efficacy of paclitaxel. The prodrug strategy alleviates the above inconveniences. For example, through the synthesis of glucuronic acid ester derivatives, it is easy to release in the presence of β-glucuronidase. In addition, 2′-ethylcarbonyl is connected to paclitaxel, which improves multidrug resistance and toxicity problems.
A considerable number of approved prodrugs attempt to solve the solubility and permeability problems of the original drug, and through this method, drug delivery and site-selective activation problems can also be improved. One of the typical cases is capecitabine, an approved oral 5-fluorouracil prodrug.
The drug was originally widely used in the anticancer treatment of solid tumors. Its main disadvantages include limited oral absorption, rapid degradation, and expected adverse drug reactions such as cytotoxicity. Capecitabine effectively solves the above problems by obtaining good oral bioavailability and selective cytotoxicity to tumor cells. After being absorbed through the intestine, it is converted into two inactive metabolites in the liver: 5’-deoxy-S-fluorocytidine (5‘-DFCR) and 5’-deoxy-S-fluorouracil (5‘-DFUR), as shown in the figure below.
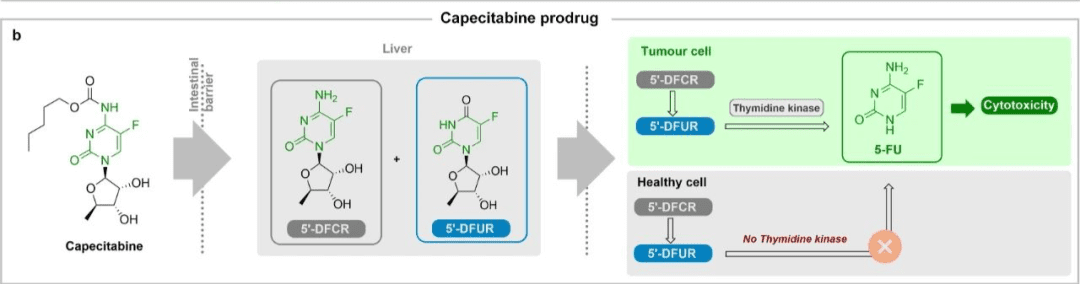
Once both metabolites enter tumor cells, they are converted into active 5-fluorouracil since thymidine kinase is highly expressed in these cells.
This targeted activation based on site-selective cytotoxicity significantly reduces 5-fluorouracil-related systemic adverse effects and improves patients’ quality of life during treatment.
In addition to site-selective activation, bioorthogonal reactions are another alternative approach to targeted therapy, in which highly efficient original drugs can be specifically activated at the tumor site. For example, the application of “click chemistry” in prodrugs, Figure a below.
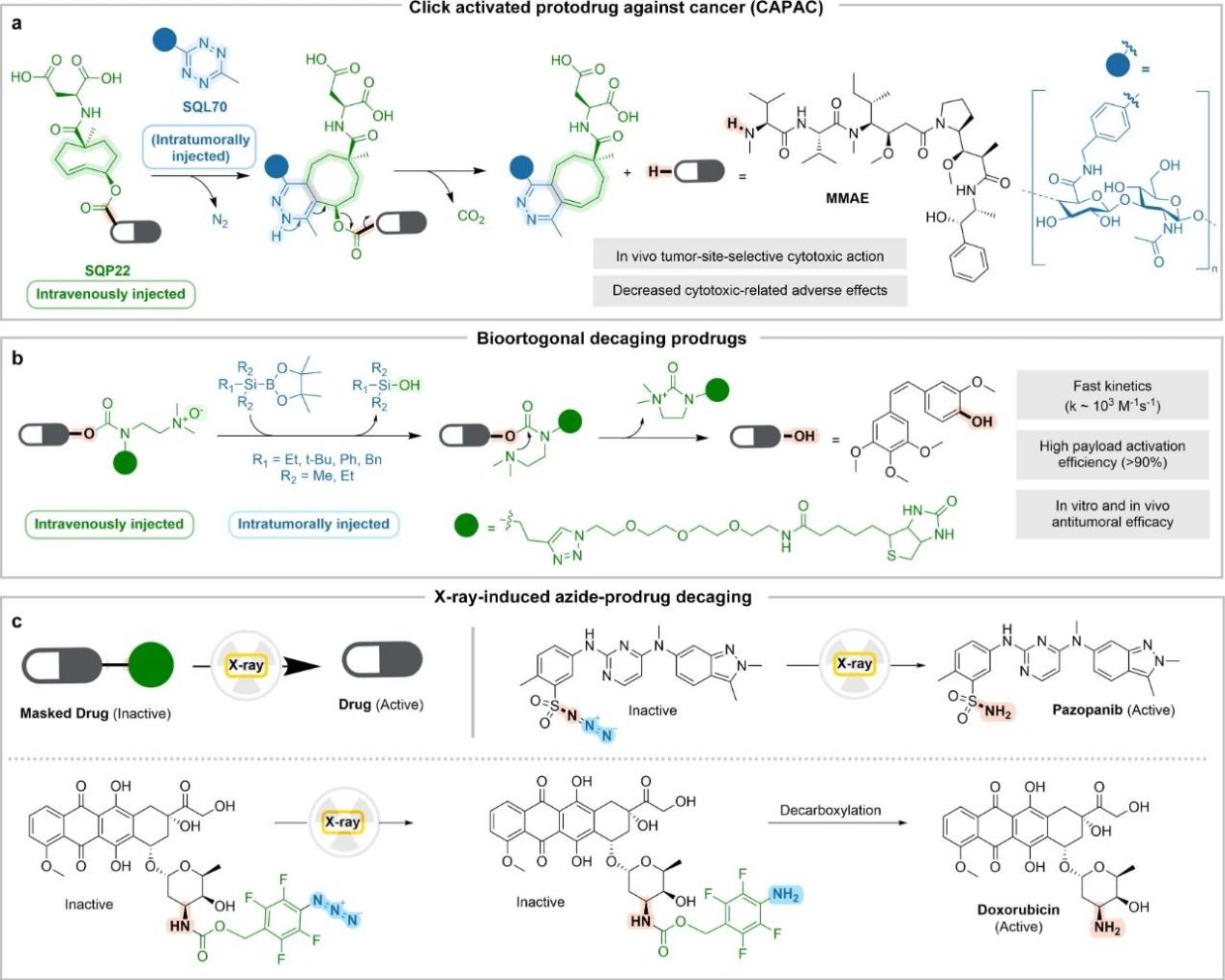
Bioorthogonal reactions ensure the stability and control of the substrate, and can achieve prodrug activation through chemical or physical stimulation.
For example, in Figure b above, the N-oxide-shielded prodrug is degraded by silborane, and several functional molecules are efficiently released. The feasibility of this chemical method has been confirmed in nuclear mice, and it can significantly inhibit tumor growth without associated toxic side effects.
For example, in Figure c above, physical stimulation under gamma/X-ray irradiation is used to activate anti-cancer therapeutic prodrugs based on pazopanib and doxorubicin in real time. The prodrug retains biological activity while being efficiently converted to the active form at clinically relevant radiation doses. This is an innovative strategy that opens up a new paradigm in local selective chemotherapy.
There are many more examples of different drug entities in prodrug applications.
For example, the problem of off-target delivery of payloads in antibody-conjugated drugs can lead to adverse effects, which can also be solved using prodrug strategies. For example, an azobenzene fragment is used in the linker of an antibody drug. Due to its high expression of azoreductase, this fragment is only cleaved in cancer cells.
Another example is when designing new drugs for the central nervous system (CNS), we face a severe challenge, namely the low permeability of the blood-brain barrier (BBB). The strategy to solve this problem is the design of prodrugs centered on fatty acid amide hydrolase (FAAH).
This enzyme is highly expressed in the central nervous system and metabolizes cannabinoid amides into carboxylates, which then use the same enzyme to cross the blood-brain barrier and enter the central nervous system. Therefore, the synthesis of amide analogues of different nuclear receptor modulators enables FAAH to successfully recognize prodrug substrates, allowing the active drug to enter the central nervous system.
2.3. Repurposing “old drugs for new uses” strategy
The strategy of repurposing old drugs, that is, repurposing approved drugs (including abandoned and withdrawn drugs) for specific indications, is one of the sources of innovation for new therapies, usually requiring a very short and affordable preclinical/clinical development period.
Strictly speaking, the strategy of repurposing old drugs does not require structural modification.
However, the authors of this article describe another possibility: the biological activity of the newly discovered drug causes synthetic changes in the active molecule, resulting in improved drugs with different indications. When the off-target effect is related to the original drug action, the change in the molecule will lead to improved activity and properties of the new drug.
The workflow of this strategy generally starts with screening a centralized drug library containing approved drugs, as shown in Figure a below. Subsequently, the initial hit is determined and evolved into a lead, which usually involves computationally guided research on the target and the selected synthetic method, either modifying the original drug or starting the synthesis again.
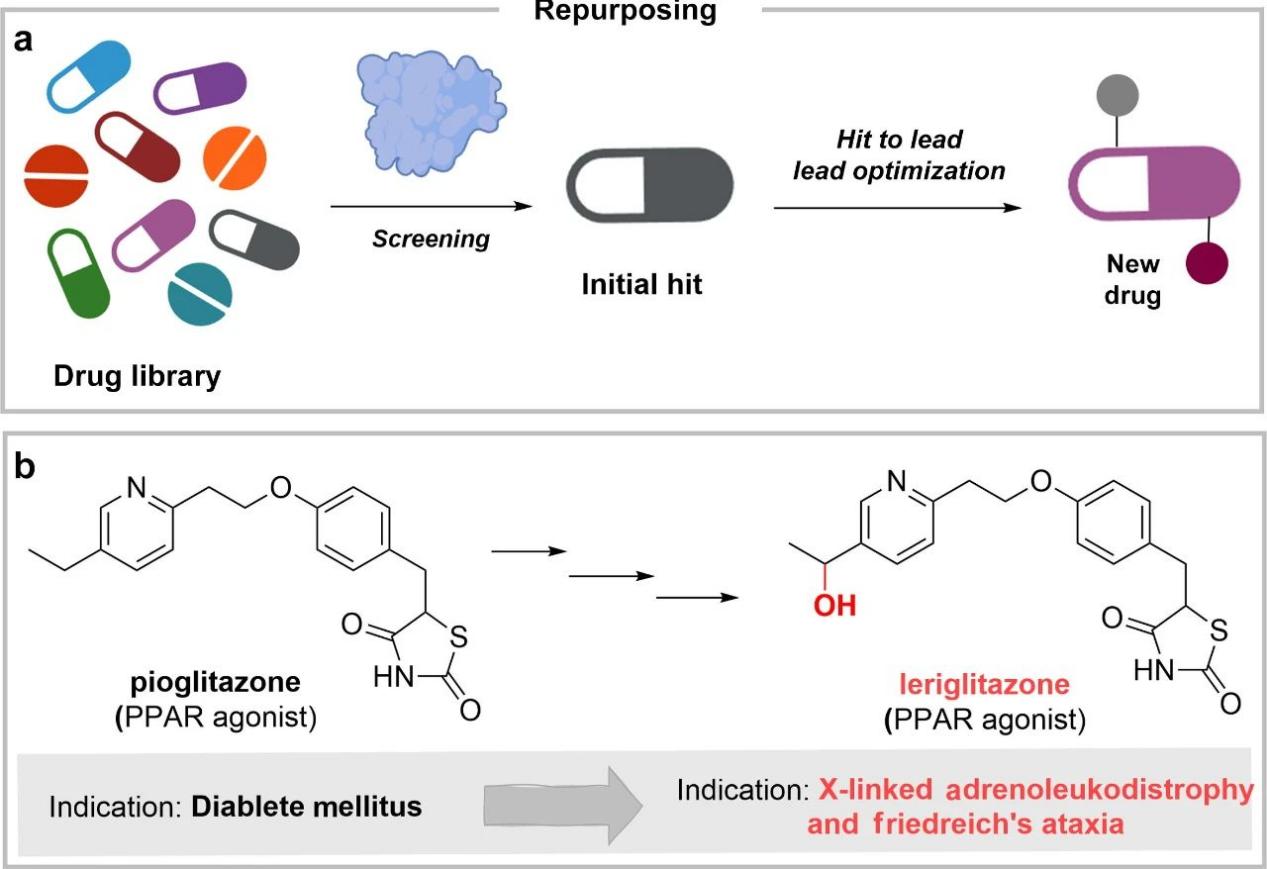
A typical example, as shown in Figure b above, is levofloxacin as a peroxisome proliferator-activated receptor (PPAR) agonist for the treatment of X-linked adrenoleukodystrophy and Friedreich’s ataxia. Through the hydroxylation of the ethyl residue, it combines effective blood-brain barrier crossing and appropriate activity at a safe dose.
In addition, computer-guided drug repurposing is becoming a reliable and effective method.
Computational methods are applicable to the docking of small molecules and the evaluation of their potential for repurposing, as well as their side effects. The programmatic output can be used to identify specific synthetic changes that help increase the potency of the tested ligand.
However, there is a limitation in the data used, which lacks updating and proper organization to improve predictions. At the same time, high-quality data should be provided in a suitable way to the currently popular algorithms such as machine learning (ML) and deep learning (DL) to realize their full potential and application in high-throughput screening.
With the emergence of quantum machine learning (QML), this strategy can be further improved in the near future, and the improvement of approved drugs obtained through quantum mechanical calculations can be influenced by artificial intelligence algorithms.
The application of QML in drug repurposing can help identify targets and fine-tune physicochemical features without extending the time scale. Furthermore, the application of QML methods in drug discovery programs will help to improve the development of this computational technology, as it will allow for an improved force field quality and better description of molecular interactions.
Summary and outlook
Approved drugs are valuable assets of biomedicine and a good starting point for new drug discovery. Discovering new drugs from drugs complements existing drug discovery methods and provides a more unique aspect for the development of new drugs.
Some applications are particularly suitable for new drug discovery based on existing drugs, such as drug modification to overcome drug resistance, interspecies differences, mutations, etc.; drug modification with suboptimal characteristics or complex synthetic pathways; and drug modification with obvious side effects.
With the emergence of new therapeutic approaches, such as dual-targeted/multi-targeted drugs, PROTACs, etc., the incorporation of existing drug fragments into larger molecular entities to play new roles is also unique to drug-based new drug discovery.
This strategy is fast and efficient, shows uniqueness in new drug discovery, and has a bright future.
Reference