Understanding the Unfolded Protein Response (UPR): A Key to Cellular Health and Disease
Abstract
The unfolded protein response (UPR) is a fundamental cellular mechanism that maintains protein homeostasis within the endoplasmic reticulum (ER). This blog post explores the activation and function of the UPR’s three primary pathways—IRE1, PERK, and ATF6—and how they collectively respond to ER stress by adjusting protein folding capacity, reducing misfolded protein accumulation, and determining cell fate. Special attention is given to the evolutionary conservation of the IRE1 pathway in yeast, the switch from adaptive responses to apoptosis, and the implications of UPR dysfunction in diseases such as cancer, neurodegeneration, and diabetes. Understanding UPR signaling not only provides insight into basic cell biology but also reveals promising targets for future therapeutic strategies.
What is the Unfolded Protein Response (UPR)?
The unfolded protein response (UPR) is a vital cellular mechanism that helps maintain protein-folding homeostasis in the endoplasmic reticulum (ER). Proteins that are destined for secretion or for membrane insertion are synthesized and folded within the ER. However, when cells experience stress—such as hypoxia, oxidative damage, viral infection, or disruptions in calcium levels—unfolded or misfolded proteins can accumulate in the ER. This buildup is referred to as ER stress, and it can threaten cell function and survival.
To combat this, cells activate the UPR, a sophisticated signaling pathway designed to restore ER homeostasis. The UPR works by reducing the burden of misfolded proteins, increasing the production of molecular chaperones that assist in protein folding, and enhancing degradation pathways that remove defective proteins. If successful, the UPR resolves the stress and normal cell function resumes. However, if ER stress is prolonged or too severe, the UPR can trigger apoptosis, or programmed cell death, to protect the organism from dysfunctional cells.
In mammals, the UPR is mediated by three main ER-resident sensor proteins: IRE1, PERK, and ATF6. These sensors detect misfolded proteins and initiate distinct but overlapping signaling cascades aimed at re-establishing ER balance. IRE1 is the most evolutionarily conserved and is found even in yeast, while PERK and ATF6 provide additional regulatory layers in higher organisms.
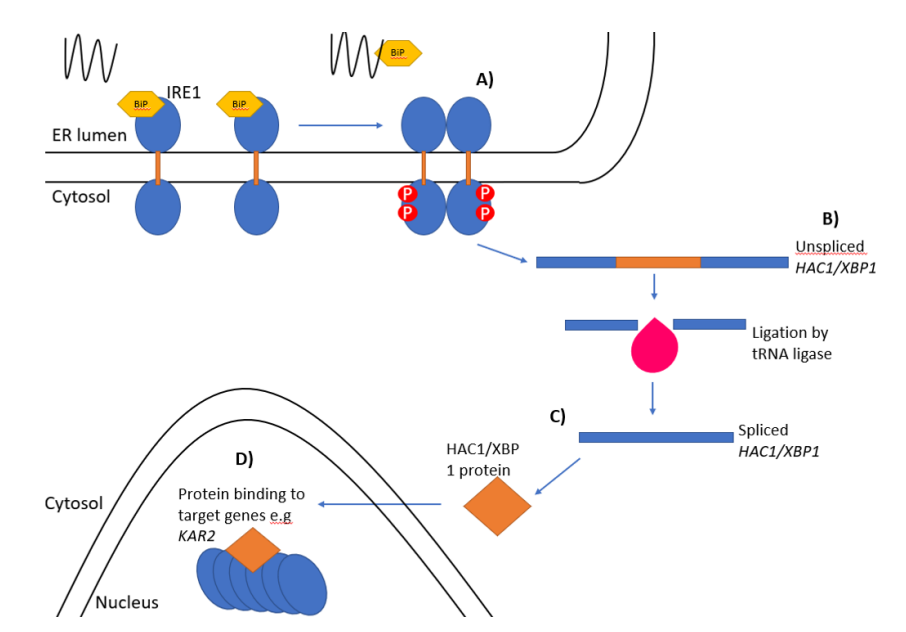
Figure 1. The unfolded protein response
The UPR is more than just a stress response; it’s a central hub for integrating signals related to metabolism, inflammation, and immune regulation. Its malfunction has been implicated in diseases such as neurodegeneration, diabetes, cancer, and inflammatory disorders. Understanding how the UPR functions—and fails—is key to developing therapeutic strategies targeting cellular stress responses.
The Three Major UPR Pathways: IRE1, PERK, and ATF6
The unfolded protein response (UPR) operates through three key signaling branches in mammalian cells: IRE1, PERK, and ATF6. Each of these transmembrane sensors is embedded in the ER membrane and plays a unique role in detecting and responding to the accumulation of unfolded proteins.
IRE1 (Inositol-Requiring Enzyme 1) is the most evolutionarily conserved of the three and has both kinase and endoribonuclease activity. Upon activation, IRE1 splices the mRNA encoding XBP1, a transcription factor that upregulates genes involved in protein folding and degradation. IRE1 also initiates RIDD (regulated IRE1-dependent decay), which degrades certain ER-localized mRNAs to reduce the protein load.
PERK (PKR-like ER Kinase) works by phosphorylating the eukaryotic initiation factor eIF2α, leading to a temporary halt in general protein translation. This gives the ER time to recover by decreasing the influx of new proteins. However, some mRNAs like ATF4 escape this repression and are preferentially translated. ATF4 then activates genes related to antioxidant responses, autophagy, and apoptosis (e.g., CHOP).
ATF6 (Activating Transcription Factor 6) follows a different route. Under stress, ATF6 translocates from the ER to the Golgi apparatus, where it is cleaved by site-1 and site-2 proteases. This cleavage releases the cytosolic portion of ATF6, which then enters the nucleus and activates genes that encode ER chaperones and components of the ER-associated degradation (ERAD) pathway.
Together, these three pathways form an integrated network. While their primary role is to restore ER function and ensure protein quality control, prolonged or excessive activation can shift the UPR from a protective response to a pro-apoptotic program.
IRE1 in Yeast and Mammals: A Closer Look
While all three UPR pathways are present in mammals, yeast cells rely solely on IRE1 to manage ER stress. This evolutionary simplification offers valuable insight into the core mechanics of the UPR.
In Saccharomyces cerevisiae (baker’s yeast), IRE1 detects unfolded proteins either directly through its luminal domain or indirectly via the ER chaperone Kar2 (homologous to BiP in mammals). Upon activation, IRE1 oligomerizes and autophosphorylates, which triggers its endoribonuclease activity. This allows it to splice HAC1 mRNA, a unique splicing process that bypasses the conventional spliceosome. The spliced HAC1 mRNA (HAC1i) is translated into a transcription factor that activates genes to alleviate ER stress.
In mammals, the orthologous process involves XBP1 mRNA splicing, which leads to the production of the active transcription factor XBP1s. This protein drives the expression of ER chaperones, ERAD components, and lipid synthesis genes. IRE1 in mammals also supports RIDD, a mechanism that degrades select ER-localized mRNAs to reduce the folding burden. Interestingly, IRE1β, found in epithelial tissues like the gut, has stronger RIDD activity compared to IRE1α.
Under persistent stress, RIDD activity can become maladaptive—targeting not only mRNAs but also microRNAs and pro-survival genes, pushing the cell toward apoptosis. This shift is known as pro-death RIDD.
The IRE1 pathway, whether in yeast or mammals, underscores the balance between adaptation and cell death in the face of ER stress. Its dual role—as a guardian of homeostasis and an initiator of cell death—makes it a compelling therapeutic target in diseases like cancer and neurodegeneration.
UPR and Cell Fate – From Adaptation to Apoptosis
While the primary goal of the unfolded protein response (UPR) is to restore balance in the endoplasmic reticulum (ER), its role extends beyond mere protein quality control. The UPR is also a critical regulator of cell fate, capable of triggering apoptosis when ER stress is unresolvable.
Initially, the UPR activates protective mechanisms: halting protein translation, boosting chaperone production, and enhancing ER-associated degradation (ERAD). These adaptive responses aim to reduce the protein folding load and resolve ER stress. However, if stress persists, the UPR initiates pro-apoptotic signaling to eliminate damaged or dysfunctional cells.
The IRE1 pathway contributes to this shift through its interaction with TRAF2 (TNF receptor-associated factor 2), which activates ASK1 (apoptosis signal-regulating kinase 1) and the JNK (c-Jun N-terminal kinase) pathway. JNK promotes apoptosis by modulating Bcl-2 family proteins and enhancing the expression of death-related genes.
Similarly, the PERK pathway leads to apoptosis through the transcription factor CHOP (C/EBP homologous protein), which is upregulated via ATF4. CHOP downregulates anti-apoptotic Bcl-2 and upregulates pro-apoptotic genes like DR5 and TRB3, tipping the balance toward cell death.
Even RIDD, which begins as a protective mRNA degradation mechanism, can contribute to apoptosis. During prolonged stress, RIDD degrades mRNAs for survival proteins and microRNAs that suppress apoptosis, further promoting cell death—a process known as pro-death RIDD.
This dual nature of the UPR—protective versus destructive—demonstrates its importance in maintaining cellular health. However, when dysregulated, it can contribute to diseases such as diabetes, neurodegeneration, and cancer. Therapeutically, modulating the UPR to enhance adaptation or block apoptosis offers promising potential.
Why UPR Matters – Disease Links and Future Directions
The unfolded protein response (UPR) is more than a stress-handling mechanism—it’s a critical component of human health. Disruptions in UPR signaling have been linked to a range of diseases, including neurodegenerative disorders, diabetes, cancer, and inflammatory conditions.
In neurodegenerative diseases such as Alzheimer’s, Parkinson’s, and ALS, protein misfolding in neurons leads to chronic ER stress. UPR pathways, particularly those involving PERK and CHOP, contribute to neuronal cell death. In diabetes, unresolved ER stress in pancreatic β-cells impairs insulin production and can lead to cell loss. The UPR is also hijacked by cancer cells, which use it to survive in harsh tumor microenvironments. Some tumors rely on UPR activity for growth and resistance to therapy, making it a potential target for anti-cancer drugs.
Furthermore, UPR dysregulation plays a role in viral infections, where viruses exploit ER stress responses to enhance replication and evade immunity. Understanding how pathogens interact with the UPR could pave the way for antiviral strategies.
Looking forward, researchers are focusing on fine-tuning UPR signaling—either boosting its adaptive capacity or blocking its pro-apoptotic effects—depending on the disease context. This could involve small molecules that inhibit IRE1 RNase activity, PERK inhibitors, or agents that enhance ER chaperone expression. However, given the complexity of UPR signaling, therapies must strike a delicate balance to avoid unwanted side effects.
Finally, the UPR’s interaction with other cellular networks—such as metabolism, autophagy, and inflammation—remains an exciting field of study. A deeper understanding of these connections could uncover new therapeutic avenues for multifactorial diseases where ER stress is just one piece of a larger puzzle.
References
Read, A., & Schröder, M. (2021). The unfolded protein response: An overview. Biology, 10(5), 384. https://doi.org/10.3390/biology10050384
Ron, D., & Walter, P. (2007). Signal integration in the endoplasmic reticulum unfolded protein response. Nature Reviews Molecular Cell Biology, 8(7), 519–529. https://doi.org/10.1038/nrm2199
Schröder, M., & Kaufman, R. J. (2005). The mammalian unfolded protein response. Annual Review of Biochemistry, 74, 739–789. https://doi.org/10.1146/annurev.biochem.73.011303.074134
Hetz, C. (2012). The unfolded protein response: Controlling cell fate decisions under ER stress and beyond. Nature Reviews Molecular Cell Biology, 13(2), 89–102. https://doi.org/10.1038/nrm3270
Hetz, C., & Papa, F. R. (2018). The unfolded protein response and cell fate control. Molecular Cell, 69(2), 169–181. https://doi.org/10.1016/j.molcel.2017.06.017
Walter, P., & Ron, D. (2011). The unfolded protein response: From stress pathway to homeostatic regulation. Science, 334(6059), 1081–1086. https://doi.org/10.1126/science.1209038
Schröder, M. (2008). Endoplasmic reticulum stress responses. Cellular and Molecular Life Sciences, 65(6), 862–894. https://doi.org/10.1007/s00018-007-7383-5
Gardner, B. M., & Walter, P. (2011). Unfolded proteins are Ire1-activating ligands that directly induce the unfolded protein response. Science, 333(6051), 1891–1894. https://doi.org/10.1126/science.1209126
Hetz, C., Zhang, K., & Kaufman, R. J. (2020). Mechanisms, regulation and functions of the unfolded protein response. Nature Reviews Molecular Cell Biology, 21(8), 421–438. https://doi.org/10.1038/s41580-020-0250-z
Hollien, J., & Weissman, J. S. (2006). Decay of endoplasmic reticulum-localized mRNAs during the unfolded protein response. Science, 313(5783), 104–107. https://doi.org/10.1126/science.1129631
Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science, 287(5453), 664–666. https://doi.org/10.1126/science.287.5453.664
Marciniak, S. J., & Ron, D. (2006). Endoplasmic reticulum stress signaling in disease. Physiological Reviews, 86(4), 1133–1149. https://doi.org/10.1152/physrev.00015.2006
Wang, M., & Kaufman, R. J. (2016). Protein misfolding in the endoplasmic reticulum as a conduit to human disease. Nature, 529(7586), 326–335. https://doi.org/10.1038/nature17041
Cláudio, N., Dalet, A., Gatti, E., & Pierre, P. (2013). Mapping the crossroads of immune activation and cellular stress response pathways. EMBO Journal, 32(9), 1214–1224. https://doi.org/10.1038/emboj.2013.80