For research use only. Not for therapeutic Use.
AM095 is a selective LPA1 receptor antagonist. The IC50 for AM095 antagonism of LPA-induced calcium flux of human or mouse LPA1-transfected CHO cells is 0.025 and 0.023 μM, respectively.
AM095 is a potent LPA1 receptor antagonist because it inhibits GTPγS binding to Chinese hamster ovary (CHO) cell membranes overexpressing recombinant human or mouse LPA1 with IC50 values of 0.98 and 0.73 μM, respectively. AM095 inhibits LPA-driven chemotaxis of CHO cells overexpressing mouse LPA1 (IC50=778 nM) and human A2058 melanoma cells (IC50=233 nM). The IC50 of AM095 in the human LPA1 GTPγS binding assay is comparable with that of our previously published compound AM966 (IC50=0.98±0.17 μM) and the Debio-0719 compound (IC50=0.60±0.04 μM)[1]. AM095 inhibits the LPA-induced calcium flux of CHO cells stably transfected with human or mouse LPA1. The IC50 for AM095 antagonism of LPA-induced calcium flux of human or mouse LPA1-transfected CHO cells is 0.025 and 0.023 μM, respectively[2].
AM095 has high oral bioavailability and a moderate half-life and is well tolerated at the doses tested in rats and dogs after oral and intravenous dosing. After oral (10 mg/kg) dosing in rats, AM095 plasma concentrations peaked at 2 h with a Cmax of 41 μM, thereafter decreasing to 10 nM by 24 h. After intravenous (2 mg/kg) dosing, a Cmax of 12 μM is observed within 15 min, which also decreased to approximately 10 nM by 24 h, yielding a t1/2 of 1.79 h. In dogs, a single oral dose of 5 mg/kg yielded a peak plasma concentration of 21 μM within 15 min of dosing, which then decreased to 10 nM by 24 h. In contrast, an intravenous dose of 2 mg/kg resulted in a Cmax of 11 μM within 15 min and decreased to 15 nM by 8 h, yielding a t1/2 of 1.5 h[1].
| CAS Number | 1345614-59-6 |
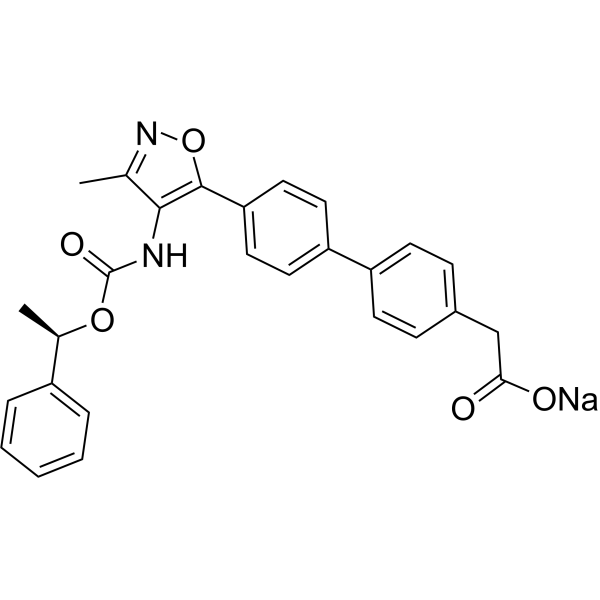
| Synonyms | sodium;2-[4-[4-[3-methyl-4-[[(1R)-1-phenylethoxy]carbonylamino]-1,2-oxazol-5-yl]phenyl]phenyl]acetate |
| Molecular Formula | C27H23N2NaO5 |
| Purity | ≥95% |
| InChI | InChI=1S/C27H24N2O5.Na/c1-17-25(28-27(32)33-18(2)20-6-4-3-5-7-20)26(34-29-17)23-14-12-22(13-15-23)21-10-8-19(9-11-21)16-24(30)31;/h3-15,18H,16H2,1-2H3,(H,28,32)(H,30,31);/q;+1/p-1/t18-;/m1./s1 |
| InChIKey | BDKDADFSIDCQGB-GMUIIQOCSA-M |
| SMILES | CC1=NOC(=C1NC(=O)OC(C)C2=CC=CC=C2)C3=CC=C(C=C3)C4=CC=C(C=C4)CC(=O)[O-].[Na+] |
| Reference | [1]. Swaney JS, et al. Pharmacokinetic and pharmacodynamic characterization of an oral lysophosphatidic acid type 1 receptor-selective antagonist. J Pharmacol Exp Ther. 2011 Mar;336(3):693-700. [2]. Castelino FV, et al. Amelioration of dermal fibrosis by genetic deletion or pharmacologic antagonism of lysophosphatidic acid receptor 1 in a mouse model of scleroderma. Arthritis Rheum. 2011 May;63(5):1405-15. |
| Chemistry Calculators | Dilution Calculator In vivo Formulation Calculator Molarity Calculator Molecular Weight Calculator Reconstitution Calculator |