For research use only. Not for therapeutic Use.
AT-56 is a selective, competitive, and highly bioavailable inhibitor of lipocalin-type prostaglandin D synthase (L-PGDS) with a Ki value of 75 µM. AT-56 inhibited human and mouse L-PGDSs in a concentration (3-250 microm)-dependent manner but did not affect the activities of hematopoietic PGD synthase (H-PGDS), cyclooxygenase-1 and -2, and microsomal PGE synthase-1. AT-56 inhibited the L-PGDS activity in a competitive manner against the substrate PGH(2) (K(m) = 14 microm) with a K(i) value of 75 microm but did not inhibit the binding of 13-cis-retinoic acid, a nonsubstrate lipophilic ligand, to L-PGDS.
| CAS Number | 162640-98-4 |
| Synonyms | 4-(5H-Dibenzo[a,d]cyclohepten-5-ylidene)-1-[4-(1H-tetrazol-5-yl)butyl]-piperidine; |
| Molecular Formula | C25H27N5 |
| Purity | ≥95% |
| Target | PGE synthase |
| Solubility | Soluble in DMSO > 10 mM |
| Storage | Store at -20C |
| IUPAC Name | 4-(dibenzo[1,2-a:1/',2/'-e][7]annulen-11-ylidene)-1-[4-(2H-tetrazol-5-yl)butyl]piperidine |
| InChI | InChI=1S/C25H27N5/c1-3-9-22-19(7-1)12-13-20-8-2-4-10-23(20)25(22)21-14-17-30(18-15-21)16-6-5-11-24-26-28-29-27-24/h1-4,7-10,12-13H,5-6,11,14-18H2,(H,26,27,28,29) |
| InChIKey | LQNGMDUIRLSESZ-UHFFFAOYSA-N |
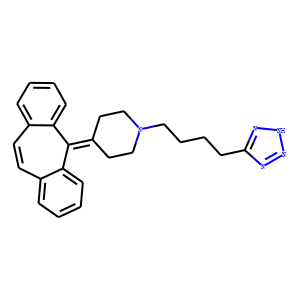
| SMILES | C1CN(CCC1=C2C3=CC=CC=C3C=CC4=CC=CC=C42)CCCCC5=NNN=N5 |
| Reference | 1:J Biol Chem. 2009 Mar 20;284(12):7623-30. doi: 10.1074/jbc.M808593200. Epub 2009 Jan 8. Biochemical, functional, and pharmacological characterization of AT-56, an orally active and selective inhibitor of lipocalin-type prostaglandin D synthase.Irikura D,Aritake K,Nagata N,Maruyama T,Shimamoto S,Urade Y, PMID: 19131342 PMCID: PMC2658056 DOI: 10.1074/jbc.M808593200 </br><span>Abstract:</span> We report here that 4-dibenzo[a,d]cyclohepten-5-ylidene-1-[4-(2H-tetrazol-5-yl)-butyl]-piperidine (AT-56) is an orally active and selective inhibitor of lipocalin-type prostaglandin (PG) D synthase (L-PGDS). AT-56 inhibited human and mouse L-PGDSs in a concentration (3-250 microm)-dependent manner but did not affect the activities of hematopoietic PGD synthase (H-PGDS), cyclooxygenase-1 and -2, and microsomal PGE synthase-1. AT-56 inhibited the L-PGDS activity in a competitive manner against the substrate PGH(2) (K(m) = 14 microm) with a K(i) value of 75 microm but did not inhibit the binding of 13-cis-retinoic acid, a nonsubstrate lipophilic ligand, to L-PGDS. NMR titration analysis revealed that AT-56 occupied the catalytic pocket, but not the retinoid-binding pocket, of L-PGDS. AT-56 inhibited the production of PGD(2) by L-PGDS-expressing human TE-671 cells after stimulation with Ca(2+) ionophore (5 microm A23187) with an IC(50) value of about 3 microm without affecting their production of PGE(2) and PGF(2alpha) but had no effect on the PGD(2) production by H-PGDS-expressing human megakaryocytes. Orally administered AT-56 (<30 mg/kg body weight) decreased the PGD(2) production to 40% in the brain of H-PGDS-deficient mice after a stab wound injury in a dose-dependent manner without affecting the production of PGE(2) and PGF(2alpha) and also suppressed the accumulation of eosinophils and monocytes in the bronco-alveolar lavage fluid from the antigen-induced lung inflammation model of human L-PGDS-transgenic mice. |
| Chemistry Calculators | Dilution Calculator In vivo Formulation Calculator Molarity Calculator Molecular Weight Calculator Reconstitution Calculator |