| Reference | 1. Drug Metab Dispos. 2013 Feb;41(2):445-56. doi: 10.1124/dmd.112.049551. Epub 2012
Nov 20.
<br>
Pharmacokinetics, metabolism, and excretion of the antidiabetic agent
ertugliflozin (PF-04971729) in healthy male subjects.
<br>
Miao Z(1), Nucci G, Amin N, Sharma R, Mascitti V, Tugnait M, Vaz AD, Callegari E,
Kalgutkar AS.
<br>
Author information: <br>
(1)Pharmacokinetics, Dynamics and Metabolism, New Chemical Entities, Groton, CT,
USA.
<br>
The disposition of ertugliflozin (PF-04971729), an orally active selective
inhibitor of the sodium-dependent glucose cotransporter 2, was studied after a
single 25-mg oral dose of [(14)C]-ertugliflozin to healthy human subjects. Mass
balance was achieved with approximately 91% of the administered dose recovered in
urine and feces. The total administered radioactivity excreted in feces and urine
was 40.9% and 50.2%, respectively. The absorption of ertugliflozin in humans was
rapid with a T(max) at ~1.0 hour. Of the total radioactivity excreted in feces
and urine, unchanged ertugliflozin collectively accounted for ~35.3% of the dose,
suggestive of moderate metabolic elimination in humans. The principal
biotransformation pathway involved glucuronidation of the glycoside hydroxyl
groups to yield three regioisomeric metabolites, M4a, M4b, and M4c (~39.3% of the
dose in urine), of which M4c was the major regioisomer (~31.7% of the dose). The
structure of M4a and M4c were confirmed to be ertugliflozin -4-O-β- and
-3-O-β-glucuronide, respectively, via comparison of the HPLC retention time and
mass spectra with authentic standards. A minor metabolic fate involved oxidation
by cytochrome P450 to yield monohydroxylated metabolites M1 and M3 and des-ethyl
ertugliflozin (M2), which accounted for ~5.2% of the dose in excreta. In plasma,
unchanged ertugliflozin and the corresponding 4-O-β- (M4a) and 3-O-β- (M4c)
glucuronides were the principal components, which accounted for 49.9, 12.2, and
24.1% of the circulating radioactivity. Overall, these data suggest that
ertugliflozin is well absorbed in humans, and eliminated largely via
glucuronidation.
<br>
2. Drug Metab Dispos. 2011 Sep;39(9):1609-19. doi: 10.1124/dmd.111.040675. Epub 2011
Jun 20.
<br>
Preclinical species and human disposition of PF-04971729, a selective inhibitor
of the sodium-dependent glucose cotransporter 2 and clinical candidate for the
treatment of type 2 diabetes mellitus.
<br>
Kalgutkar AS(1), Tugnait M, Zhu T, Kimoto E, Miao Z, Mascitti V, Yang X, Tan B,
Walsky RL, Chupka J, Feng B, Robinson RP.
<br>
Author information: <br>
(1)Pharmacokinetics, Dynamics, and Metabolism Department, Pfizer Global Research
and Development, Groton, CT 06340, USA. [email protected]
<br>
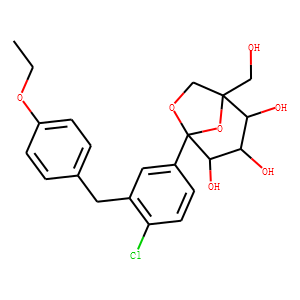
(1S,2S,3S,4R,5S)-5-[4-Chloro-3-(4-ethoxybenzyl)phenyl]-1-hydroxymethyl-6,8-dioxab
icyclo[3.2.1]octane-2,3,4-triol (PF-04971729), a potent and selective inhibitor
of the sodium-dependent glucose cotransporter 2, is currently in phase 2 trials
for the treatment of diabetes mellitus. This article describes the preclinical
species and in vitro human disposition characteristics of PF-04971729 that were
used in experiments performed to support the first-in-human study. Plasma
clearance was low in rats (4.04 ml · min(-1) · kg(-1)) and dogs (1.64 ml ·
min(-1) · kg(-1)), resulting in half-lives of 4.10 and 7.63 h, respectively.
Moderate to good bioavailability in rats (69%) and dogs (94%) was observed after
oral dosing. The in vitro biotransformation profile of PF-04971729 in liver
microsomes and cryopreserved hepatocytes from rat, dog, and human was
qualitatively similar; prominent metabolic pathways included monohydroxylation,
O-deethylation, and glucuronidation. No human-specific metabolites of PF-04971729
were detected in in vitro studies. Reaction phenotyping studies using recombinant
enzymes indicated a role of CYP3A4/3A5, CYP2D6, and UGT1A9/2B7 in the metabolism
of PF-04971729. No competitive or time-dependent inhibition of the major human
cytochrome P450 enzymes was discerned with PF-04971729. Inhibitory effects
against the organic cation transporter 2-mediated uptake of [(14)C]metformin by
PF-04971729 also were very weak (IC(50) = ~900 μM). Single-species allometric
scaling of rat pharmacokinetics of PF-04971729 was used to predict human
clearance, distribution volume, and oral bioavailability. Human pharmacokinetic
predictions were consistent with the potential for a low daily dose.
First-in-human studies after oral administration indicated that the human
pharmacokinetics/dose predictions for PF-04971729 were in the range that is
likely to yield a favorable pharmacodynamic response.
<br>
|