| Reference | 1. Angew Chem Int Ed Engl. 2016 Mar 7;55(11):3590-5. doi: 10.1002/anie.201509240.
Epub 2016 Feb 16.
<br>
Conformational Studies and Atropisomerism Kinetics of the ALK Clinical Candidate
Lorlatinib (PF-06463922) and Desmethyl Congeners.
<br>
Elleraas J(1), Ewanicki J(1), Johnson TW(1), Sach NW(1), Collins MR(2),
Richardson PF(3).
<br>
Author information: <br>
(1)Oncology Medicinal Chemistry, Pfizer, La Jolla, 10770 Science Center Drive,
San Diego, CA, 92121, USA.
(2)Oncology Medicinal Chemistry, Pfizer, La Jolla, 10770 Science Center Drive,
San Diego, CA, 92121, USA. [email protected].
(3)Oncology Medicinal Chemistry, Pfizer, La Jolla, 10770 Science Center Drive,
San Diego, CA, 92121, USA. [email protected].
<br>
Lorlatinib (PF-06463922) is an ALK/ROS1 inhibitor and is in clinical trials for
the treatment of ALK positive or ROS1 positive NSCLC (i.e. specific subsets of
NSCLC). One of the laboratory objectives for this molecule indicated that it
would be desirable to advance a molecule which was CNS penetrant in order to
treat brain metastases. From this perspective, a macrocyclic template was
attractive for a number of reasons. In particular, this template reduces the
number of rotatable bonds, provides the potential to shield polar surface area
and reinforces binding through a restricted conformation. All of these features
led to better permeability for the molecules of interest and thus increased the
chance for better blood brain barrier penetration. With a CNS penetrant molecule,
kinase selectivity is a key consideration particularly with regard to proteins
such as TrkB, which are believed to influence cognitive function. Removal of the
chiral benzylic methyl substituent from lorlatinib was perceived as not only a
means to simplify synthetic complexity, but also as a strategy to further
truncate the molecule of interest. Examination of the NMR of the desmethyl
analogues revealed that the compound existed as a mixture of atropisomers, which
proved separable by chiral SFC. The individual atropisomers were evaluated
through a series of in vitro assays, and shown to have a favorable selectivity
profile when compared to lorlatinib. The challenge to develop such a molecule
lies in the rate at which the atropisomers interchange dictated by the energy
barrier required to do this. Here, we describe the synthesis of the desmethyl
macrocycles, conformational studies on the atropisomers, and the kinetics of the
interconversion. In addition, the corresponding conformational studies on
lorlatinib are reported providing a hypothesis for why a single diastereomer is
observed when the chiral benzylic methyl group is introduced.
<br>
2. Oncotarget. 2015 Mar 20;6(8):5720-34.
<br>
NPM/ALK mutants resistant to ASP3026 display variable sensitivity to alternative
ALK inhibitors but succumb to the novel compound PF-06463922.
<br>
Mologni L(1), Ceccon M(1), Pirola A(1), Chiriano G(2), Piazza R(1), Scapozza
L(2), Gambacorti-Passerini C(1)(3).
<br>
Author information: <br>
(1)University of Milano-Bicocca, Dept. of Health Sciences, Monza, Italy.
(2)University of Geneva, School of Pharmaceutical Sciences, Geneva, Switzerland.
(3)San Gerardo Hospital, Hematology Unit, Monza, Italy.
<br>
ALK is involved in the onset of several tumors. Crizotinib (XalkoriTM), a potent
ALK inhibitor, represents the current front-line treatment for ALK+ NSCLC and
shows great clinical efficacy. However, resistant disease often develops after
initial response. ASP3026 is a novel second-generation ALK inhibitor with
activity on crizotinib-resistant ALK-L1196M gatekeeper mutant. As resistance is
likely to be a relevant hurdle for any drug, we sought to determine the
resistance profile of ASP3026 in the context of NPM/ALK+ ALCL. We selected six
ASP3026-resistant cell lines by culturing human ALCL cells in the presence of
increasing concentrations of drug. The established resistant cell lines carry
several point mutations in the ALK kinase domain (G1128S, C1156F, I1171N/T,
F1174I, N1178H, E1210K and C1156F/D1203N were the most frequent) that are shown
to confer resistance to ASP3026 in the Ba/F3 cell model. All mutants were
profiled for cross-resistance against a panel of clinically relevant inhibitors
including ceritinib, alectinib, crizotinib, AP26113 and PF-06463922. Finally, a
genetically heterogeneous ASP3026-resistant cell line was exposed to second-line
treatment simulations with all inhibitors. The population evolved according to
relative sensitivity of its mutant subclones to the various drugs. Compound
PF-06463922 did not allow the outgrowth of any resistant clone, at non-toxic
doses.
<br>
3. J Med Chem. 2014 Jun 12;57(11):4720-44. doi: 10.1021/jm500261q. Epub 2014 Jun 3.
<br>
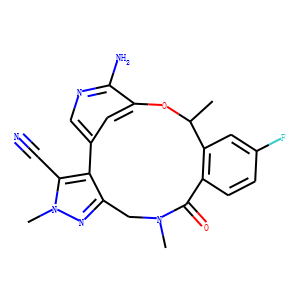
Discovery of
(10R)-7-amino-12-fluoro-2,10,16-trimethyl-15-oxo-10,15,16,17-tetrahydro-2H-8,4-(m
etheno)pyrazolo[4,3-h][2,5,11]-benzoxadiazacyclotetradecine-3-carbonitrile
(PF-06463922), a macrocyclic inhibitor of anaplastic lymphoma kinase (ALK) and
c-ros oncogene 1 (ROS1) with preclinical brain exposure and broad-spectrum
potency against ALK-resistant mutations.
<br>
Johnson TW(1), Richardson PF, Bailey S, Brooun A, Burke BJ, Collins MR, Cui JJ,
Deal JG, Deng YL, Dinh D, Engstrom LD, He M, Hoffman J, Hoffman RL, Huang Q,
Kania RS, Kath JC, Lam H, Lam JL, Le PT, Lingardo L, Liu W, McTigue M, Palmer CL,
Sach NW, Smeal T, Smith GL, Stewart AE, Timofeevski S, Zhu H, Zhu J, Zou HY,
Edwards MP.
<br>
Author information: <br>
(1)La Jolla Laboratories, Pfizer Worldwide Research and Development , 10770
Science Center Drive, San Diego, California 92121, United States.
<br>
Although crizotinib demonstrates robust efficacy in anaplastic lymphoma kinase
(ALK)-positive non-small-cell lung carcinoma patients, progression during
treatment eventually develops. Resistant patient samples revealed a variety of
point mutations in the kinase domain of ALK, including the L1196M gatekeeper
mutation. In addition, some patients progress due to cancer metastasis in the
brain. Using structure-based drug design, lipophilic efficiency, and
physical-property-based optimization, highly potent macrocyclic ALK inhibitors
were prepared with good absorption, distribution, metabolism, and excretion
(ADME), low propensity for p-glycoprotein 1-mediated efflux, and good passive
permeability. These structurally unusual macrocyclic inhibitors were potent
against wild-type ALK and clinically reported ALK kinase domain mutations.
Significant synthetic challenges were overcome, utilizing novel transformations
to enable the use of these macrocycles in drug discovery paradigms. This work led
to the discovery of 8k (PF-06463922), combining broad-spectrum potency, central
nervous system ADME, and a high degree of kinase selectivity.
<br>
|