| Reference | 1. Invest New Drugs. 2008 Feb;26(1):59-65. Epub 2007 Oct 16.
<br>
A phase 1 study of SNS-032 (formerly BMS-387032), a potent inhibitor of
cyclin-dependent kinases 2, 7 and 9 administered as a single oral dose and weekly
infusion in patients with metastatic refractory solid tumors.
<br>
Heath EI(1), Bible K, Martell RE, Adelman DC, Lorusso PM.
<br>
Author information: <br>
(1)Wayne State University/Karmanos Cancer Institute, 4100 John R, 4HWCRC,
Detroit, MI 48201, USA. [email protected]
<br>
PURPOSE: SNS-032, (formerly BMS-387032) is a potent and selective inhibitor of
cyclin-dependent kinases (CDK) 2, 7 and 9. The primary objective of the study was
to establish the maximum tolerated dose (MTD), the maximum administered dose
(MAD), dose limiting toxicity (DLT), and the recommended phase 2 dose for SNS-032
when administered as a weekly 1-h infusion. The secondary objective was to assess
the safety and tolerability of SNS-032 and to evaluate its bioavailability as an
oral solution.<br>
METHODS: Patients with metastatic solid tumors or refractory lymphoma were
treated with a starting dose of 4 mg/m2 intravenously administered over 1-h with
a cycle defined as 3 weekly doses of SNS-032 every 21 days. Three patient cohorts
were utilized in the dose-escalation schema. Pharmacokinetic studies were
performed. For the 13 and 16 mg/m2 dose cohorts, the first dose of cycle 2 was
given as an oral solution to estimate the oral bioavailability of the drug in
humans.<br>
RESULTS: A total of 21 patients were enrolled. Twenty treated patients received a
total of 39 cycles of treatment. The most common treatment-related adverse events
occurring with greater than 20% incidence were fatigue (25%) and nausea (20%).
Following intravenous administration, plasma concentrations declined in a
biphasic manner, resulting in mean terminal half-lives between 5 and 10 hours.
The mean Cmax and AUC0-inf increased nearly linearly with dose, ranging from
0.067 to 0.287 microg/ml and 0.103 to 0.553 microg h/ml, respectively. The CL and
Vss remained unchanged with increasing dose levels, averaging 38 l/h/m2 and 212
l/m2, respectively. Average oral bioavailability was 19% (range: 4-33%). Three
(15%) patients experienced a best response of stable disease. Study enrollment
was terminated during dose-escalation due to a change in the development strategy
for the study drug.<br>
CONCLUSIONS: SNS-032 administered as a weekly 1-h infusion was well tolerated,
although study enrollment was terminated during dose-escalation and the MTD of
SNS-032 administered intravenously on days 1, 8, and 15 of each treatment cycle
was not reached. Tumor progression or stable disease was determined to be the
best response in all evaluable patients. At the dose levels tested, the oral
bioavailability of SNS-032 ranged from 4-33%. The data suggest that oral
administration of SNS-032 may be feasible, though the tolerability and
bioavailability of the oral formulation would have to be formally assessed.
<br>
2. Cancer Chemother Pharmacol. 2005 Feb;55(2):110-6. Epub 2004 Aug 27.
<br>
P-glycoprotein plays a role in the oral absorption of BMS-387032, a potent
cyclin-dependent kinase 2 inhibitor, in rats.
<br>
Kamath AV(1), Chong S, Chang M, Marathe PH.
<br>
Author information: <br>
(1)Department of Metabolism and Pharmacokinetics, Bristol-Myers Squibb
Pharmaceutical Research Institute, Princeton, NJ 08543, USA.
[email protected]
<br>
PURPOSE: BMS-387032, a novel cyclin-dependent kinase 2 inhibitor, is currently in
phase I clinical trials for anticancer therapy. The oral bioavailability of
BMS-387032 has been found to be about 31% in rats. Absorption and first-pass
metabolism were evaluated as possible reasons for the incomplete oral
bioavailability in rats.<br>
METHODS: Male Sprague-Dawley rats were given single doses of BMS-387032
intraarterially (9.1 mg/kg), orally (9.1 mg/kg), or intraportally (10 mg/kg). The
routes of excretion of BMS-387032 after intravenous dosing were investigated in
bile-duct-cannulated rats. The rate of metabolism of BMS-387032 was investigated
in liver microsomes. The permeability of BMS-387032 was evaluated using Caco-2
cells, an in vitro model of the intestinal epithelium. To determine if BMS-387032
was a P-glycoprotein substrate, brain uptake studies were conducted in
P-glycoprotein knockout versus wildtype mice.<br>
RESULTS: The exposure in rats after an intraportal dose was similar to that after
an intraarterial dose, indicating that absorption may play a greater role than
liver first-pass metabolism in the low oral bioavailability seen in rats. After
an intravenous dose, the percent of dose excreted unchanged in the urine and bile
over a 9-h period was 28% and 11%, respectively. In vitro studies in rat liver
microsomes showed low rates of metabolism of BMS-387032. The Caco-2 cell
permeability of BMS-387032 was <15 nm/s in the apical to basolateral direction,
and 161 nm/s in the basolateral to apical direction, indicating that it may be a
substrate for an intestinal efflux transporter. A P-glycoprotein binding assay
showed that BMS-387032 might be a P-glycoprotein modulator. Brain penetration
studies in mice showed brain levels of BMS-387032 about 3.5-fold higher in
P-glycoprotein knockout mice than in wildtype mice, providing evidence of
BMS-387032 being a P-glycoprotein substrate.<br>
CONCLUSIONS: Poor absorption may be playing a greater role than extensive
first-pass metabolism in the incomplete oral bioavailability of BMS-387032 seen
in rats. The efflux transporter, P-glycoprotein, may be responsible for limiting
absorption, as BMS-387032 appears to be a substrate of P-glycoprotein.
<br>
3. J Med Chem. 2004 Mar 25;47(7):1719-28.
<br>
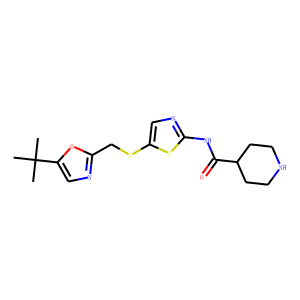
N-(cycloalkylamino)acyl-2-aminothiazole inhibitors of cyclin-dependent kinase 2.
N-[5-[[[5-(1,1-dimethylethyl)-2-oxazolyl]methyl]thio]-2-thiazolyl]-4-
piperidinecarboxamide (BMS-387032), a highly efficacious and selective antitumor
agent.
<br>
Misra RN(1), Xiao HY, Kim KS, Lu S, Han WC, Barbosa SA, Hunt JT, Rawlins DB, Shan
W, Ahmed SZ, Qian L, Chen BC, Zhao R, Bednarz MS, Kellar KA, Mulheron JG,
Batorsky R, Roongta U, Kamath A, Marathe P, Ranadive SA, Sack JS, Tokarski JS,
Pavletich NP, Lee FY, Webster KR, Kimball SD.
<br>
Author information: <br>
(1)Bristol-Myers Squibb Pharmaceutical Research Institute, PO Box 4000,
Princeton, New Jersey 08543-4000, USA. [email protected]
<br>
Erratum in
J Med Chem. 2015 Sep 24;58(18):7609.
<br>
N-Acyl-2-aminothiazoles with nonaromatic acyl side chains containing a basic
amine were found to be potent, selective inhibitors of CDK2/cycE which exhibit
antitumor activity in mice. In particular, compound 21
[N-[5-[[[5-(1,1-dimethylethyl)-2-oxazolyl]methyl]thio]-2-thiazolyl]-4-piperidinec
arboxamide, BMS-387032], has been identified as an ATP-competitive and
CDK2-selective inhibitor which has been selected to enter Phase 1 human clinical
trials as an antitumor agent. In a cell-free enzyme assay, 21 showed a CDK2/cycE
IC(50) = 48 nM and was 10- and 20-fold selective over CDK1/cycB and CDK4/cycD,
respectively. It was also highly selective over a panel of 12 unrelated kinases.
Antiproliferative activity was established in an A2780 cellular cytotoxicity
assay in which 21 showed an IC(50) = 95 nM. Metabolism and pharmacokinetic
studies showed that 21 exhibited a plasma half-life of 5-7 h in three species and
moderately low protein binding in both mouse (69%) and human (63%) serum. Dosed
orally to mouse, rat, and dog, 21 showed 100%, 31%, and 28% bioavailability,
respectively. As an antitumor agent in mice, 21 administered at its
maximum-tolerated dose exhibited a clearly superior efficacy profile when
compared to flavopiridol in both an ip/ip P388 murine tumor model and in a
s.c./i.p. A2780 human ovarian carcinoma xenograft model.
<br>
|