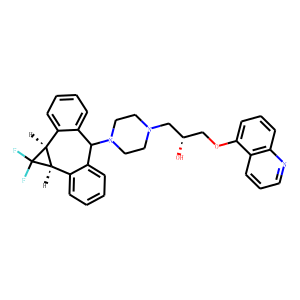
| Synonyms | Zosuquidar; (2R)-1-(4-((1aR,10bS)-1,1-difluoro-1,1a,6,10b-tetrahydrodibenzo[a,e]cyclopropa[c][7]annulen-6-yl)piperazin-1-yl)-3-(quinolin-5-yloxy)propan-2-ol
|
| InChI | InChI=1S/C32H31F2N3O2/c33-32(34)29-22-7-1-3-9-24(22)31(25-10-4-2-8-23(25)30(29)32)37-17-15-36(16-18-37)19-21(38)20-39-28-13-5-12-27-26(28)11-6-14-35-27/h1-14,21,29-31,38H,15-20H2/t21-,29-,30+,31?/m1/s1 |
| Reference | 1. Cancer Chemother Pharmacol. 2004 Feb;53(2):173-8. Epub 2003 Nov 7.
<br>
The influence of the P-glycoprotein inhibitor zosuquidar trihydrochloride
(LY335979) on the brain penetration of paclitaxel in mice.
<br>
Kemper EM(1), Cleypool C, Boogerd W, Beijnen JH, van Tellingen O.
<br>
Author information: <br>
(1)Department of Clinical Chemistry, The Netherlands Cancer Institute/Antoni van
Leeuwenhoek Huis, Plesmanlaan 121, 1066 CX Amsterdam, The Netherlands.
<br>
We determined the effect of zosuquidar.3HCl, an inhibitor of P-gp, on the
penetration of the anticancer drug paclitaxel into the brain. Zosuquidar.3HCl was
administered orally at 25 and 80 mg/kg 1 h before i.v. paclitaxel and i.v. at 20
mg/kg 10 min and 1 h before paclitaxel. The concentrations of paclitaxel in
plasma and tissues and of zosuquidar.3HCl in plasma were quantified by
high-performance liquid chromatography. The results revealed 3.5-fold and 5-fold
higher paclitaxel levels in the brain of wild-type mice treated orally with 25
and 80 mg/kg zosuquidar.3HCl, respectively. However, complete inhibition as in
P-gp knockout mice (11-fold increase) was not achieved. Zosuquidar.3HCl also
increased the paclitaxel concentrations in plasma and tissues to levels similar
to those observed in P-gp knockout mice, suggesting selective P-gp inhibition of
zosuquidar.3HCl. When zosuquidar.3HCl was administered i.v. 10 min before
paclitaxel, the paclitaxel levels in the brain of wild-type mice increased by
5.6-fold, whereas the increase was only 2.1-fold when zosuquidar.3HCl was
administered 1 h before paclitaxel. This suggests that the inhibition of P-gp at
the blood-brain barrier by zosuquidar.3HCl is rapidly reversible and that the
concentrations of zosuquidar.3HCl in the plasma have already declined to levels
insufficient to inhibit P-gp at the blood-brain barrier. In conclusion,
zosuquidar.3HCl is only moderately active as an inhibitor of P-gp at the
blood-brain barrier.
<br>
2. J Chromatogr B Analyt Technol Biomed Life Sci. 2003 Dec 5;798(1):63-8.
<br>
Bioanalysis of zosuquidar trihydrochloride (LY335979) in small volumes of human
and murine plasma by ion-pairing reversed-phase high-performance liquid
chromatography.
<br>
Kemper EM(1), Ouwehand M, Beijnen JH, van Tellingen O.
<br>
Author information: <br>
(1)Department of Clinical Chemistry, The Netherlands Cancer Institute/Antoni van
Leeuwenhoek Huis, Plesmanlaan 121, 1066 CX Amsterdam, The Netherlands.
<br>
We have developed and validated a sensitive and selective method for the
quantitative determination of the P-glycoprotein inhibitor zosuquidar (LY335979)
in human and murine plasma using only 50 microl sample volumes. Sample
pretreatment involved liquid-liquid extraction with tert-butyl methyl ether.
Zosuquidar and the internal standard chlorpromazine were separated using a narrow
bore column (2.1 mm x 150 mm) packed with 3.5 microm symmetry C(18) material. The
mobile phase consisted of 38% (v/v) acetonitrile in 50mM ammonium acetate buffer
pH 3.8 containing 0.005 M 1-octyl sulfonic acid and was delivered at 0.2 ml/min.
Detection was performed with a fluorescence detector set at an excitation
wavelength of 260 nm and an emission wavelength of 460 nm. The calibration curve
was prepared in blank human plasma and was linear over the dynamic range (10-1000
ng/ml). The lower limit of quantitation was 20 ng/ml. The validation results
showed that the assay was selective and reproducible. Within the range of the
calibration curve the accuracy was close to 100% and within-day and between-day
precision were within the generally accepted 15% range. This method was applied
to study the pharmacokinetics of i.v. administered zosuquidar in mice. The
sensitivity of the assay was sufficient to determine the drug concentration in
plasma samples obtained up to 24 h after administration.
<br>
|